Somatic SNV and Indel Manual Review
Somatic Variant Refinement
Automated pipelines can identify and filter many false variant calls that result from sequencing errors, misalignment of reads, and other factors; however, additional refinement of somatic variants is often required to eliminate variant caller inaccuracies. Following automated variant calling, heuristic filtering and manual review of aligned read sequences is required to identify a high-quality list of somatic variants. This process of filtering and manual review is deemed somatic variant refinement. Here we describe methods to filter the somatic variant identified by mutect, varscan, and strelka for chromosome 6 and 17. First we filter the VCF file using VAF, total coverage, and variant coverage. We subsequently describe the Integrative Genomics Viewer (IGV) and IGVNavigator, which can be used to standardize and streamline manual review.
Somatic Variant Filtering
Somatic variant filtering requires setting heuristic cutoffs for various sequencing data to eliminate called variants that might be false positives. This requires first defining the thresholds and then employing these thresholds on variant files. Typically we set thresholds for variant allele fraction (VAF), total coverage, and variant coverage.
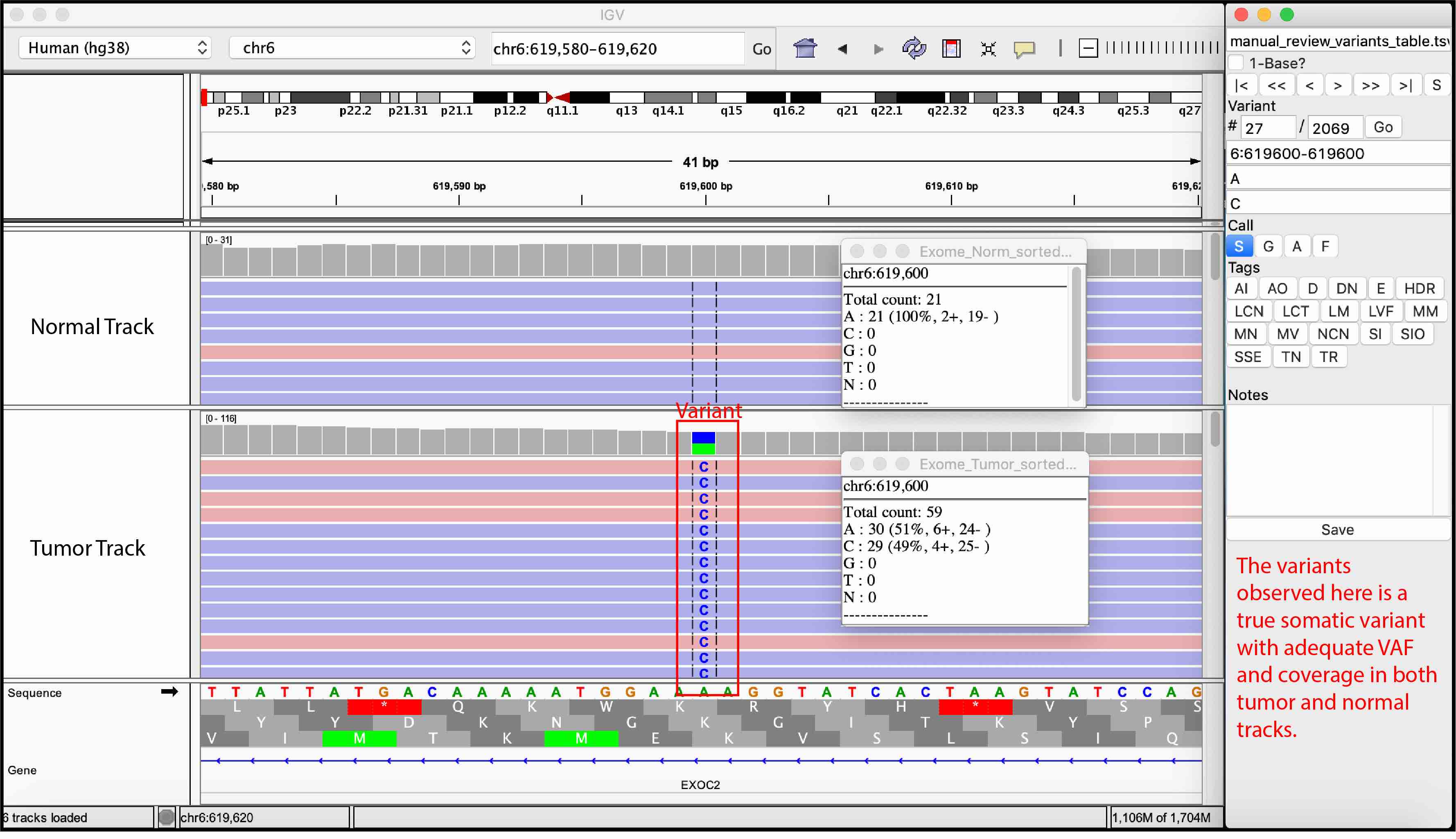
Variant allele fraction (VAF): The VAF represents the relative frequency of an allele at a particular locus, which serves as a proxy for number of cells containing the variant (i.e., 5% VAF means that 10% of the cells in the sample contain the variant). Typically, at least 5% VAF is required to make confident calls (given 20X coverage); however, this threshold is experiment-specific. For experiments with higher average coverage, the minimum VAF threshold can be reduced accordingly.
Total coverage: The total coverage indicates the number of sequencing reads that align to the locus of interest. Typically, at least 20X coverage in both normal and tumor tracks is required to make accurate calls. The rationale for the normal track coverage threshold is that if a sequencing artifact is present at a relatively low frequency (<5% occurrence), and if the normal track has <20 reads, it is difficult to confidently rule out the presence of a sequencing artifact. Additionally, calling a variant with low coverage has important downstream implications. When the tumor track has low coverage, variant allele frequency (VAF) estimates can be heavily influence by sequencing noise and sampling bias. This may result in: a false negative with an underestimated VAF, a false positive due to over-estimation of the VAF, and/or a true positive call with inaccurate VAF.
Variant coverage: The variant coverage indicates the number of sequencing reads aligned to the locus of interest that contain the variant. Typically, we require at least 5 variant-supporting reads to confidently call a variant as somatic. This requirement eliminates short insert variants, which occur if the variant is found on small nucleic acid fragments whereby sequencing from each end results in overlapping reads. Variants supported by read pairs produced from these short fragments can result in the appearance of two independent reads supporting a variant when in reality, they represent only a single nucleic acid molecule. Using variant coverage thresholds eliminate many variants that require manual review but it can unfortunately eliminate true somatic variants that are sub-clonal and only found in a few cells.
Manual Review
After somatic variant filtering, manual review of aligned read sequences is required to identify a high-quality list of somatic variants. Manual review allows individuals to incorporate information not considered by automated variant callers. For example, a trained eye can discern misclassifications attributable to overlapping errors at the ends of sequence reads, preferential amplification of smaller fragments, or poor alignment in areas of low complexity. A standard operating procedure described by our lab can be used to better understand how to setup and conduct manual review [INSERT LINK].
Briefly, manual review requires use of a genomic viewer, a BAM file of aligned sequencing reads, and a BED or BED-like file for all variant that require manual review. Each variant can be observed in the genomic viewer and variants can be labeled as true somatic variants, false somatic variants, germline variants, or ambiguous. After manual review, only variants that are labeled as true somatic variants are subsequently used for annotation.
For convenience, we provided examples of true somatic variants and false positives observed during manual review of our sequencing data.
Somatic Variant
False Variant
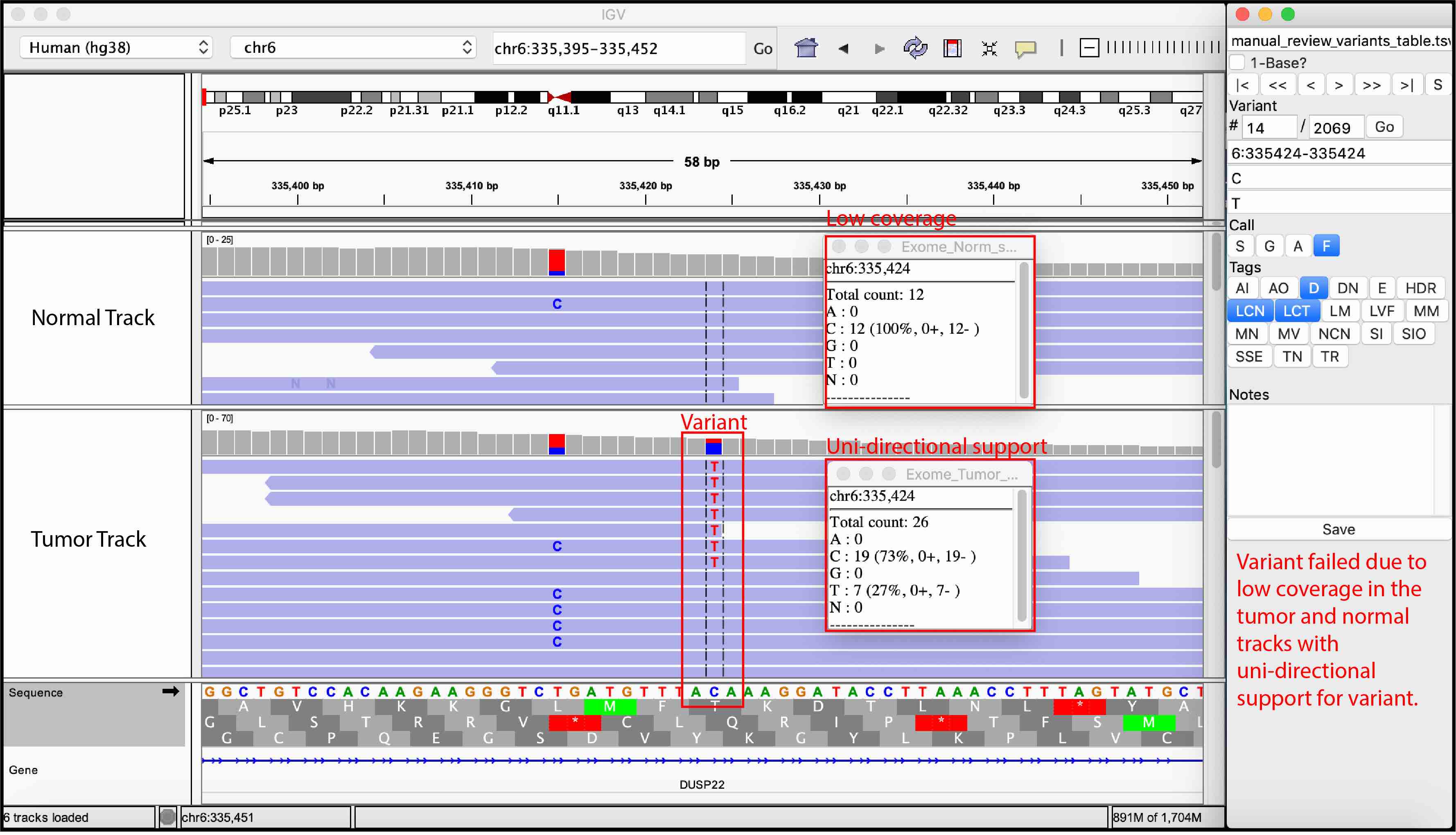
False Variant
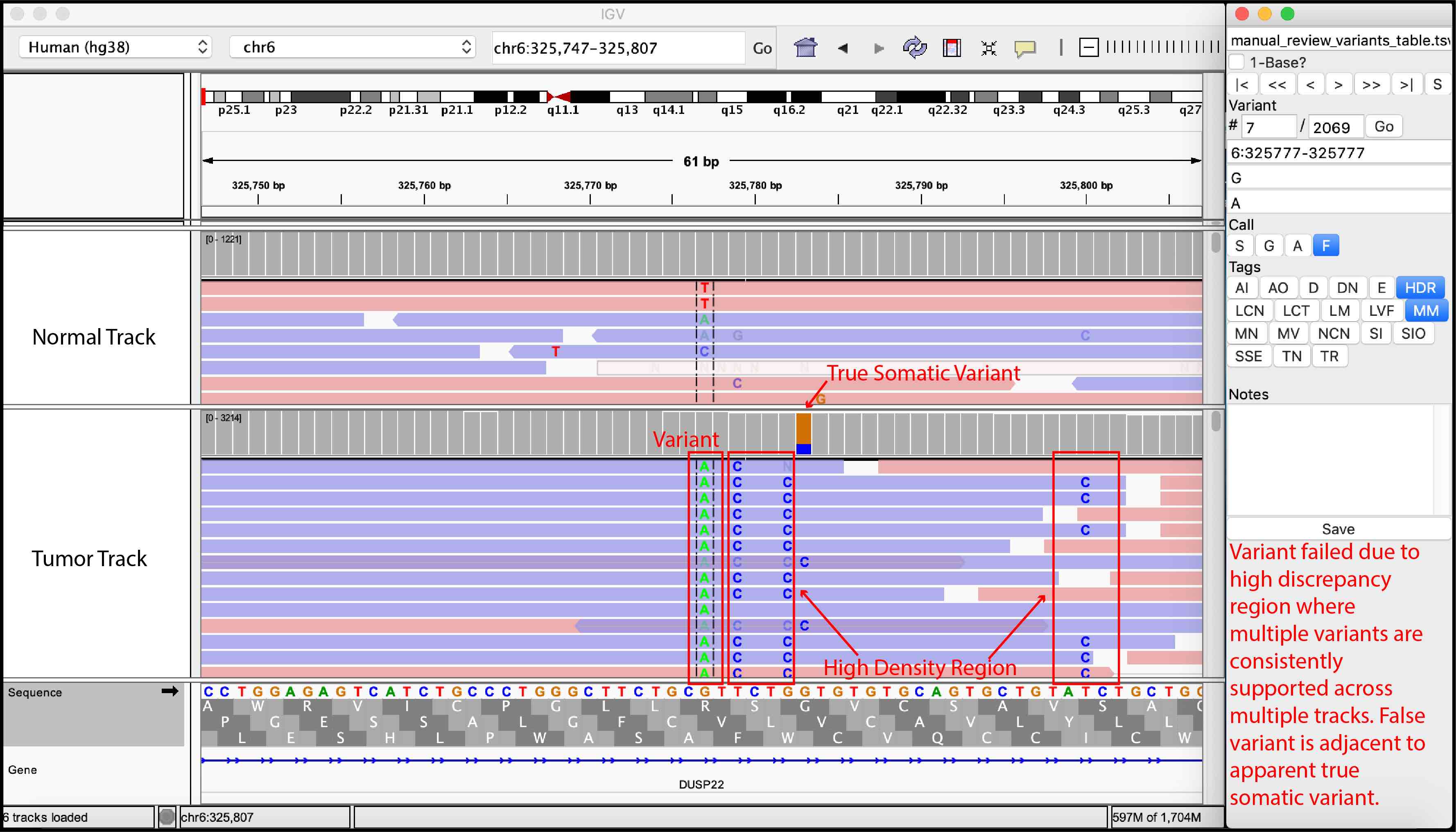
False Variant
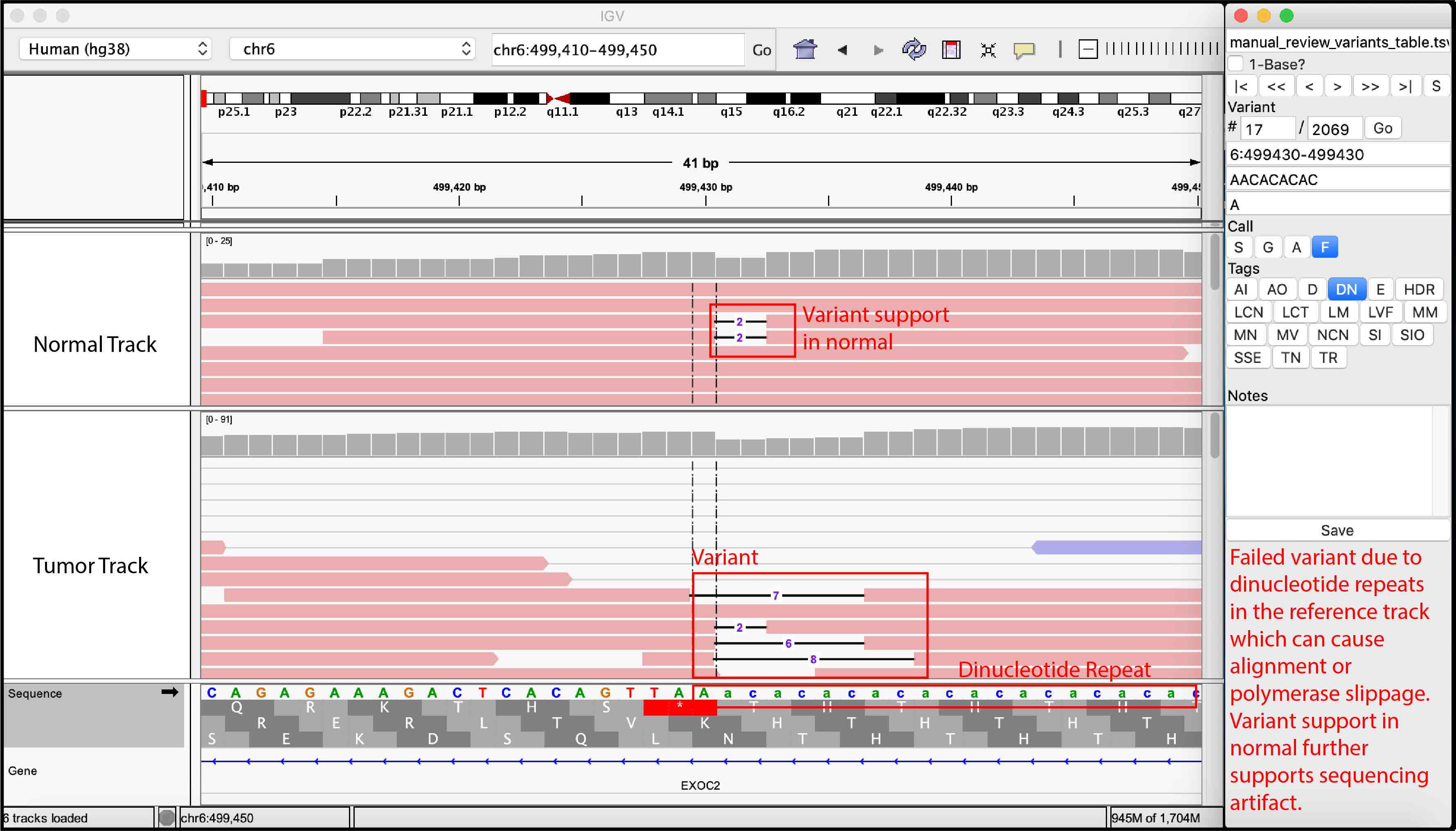
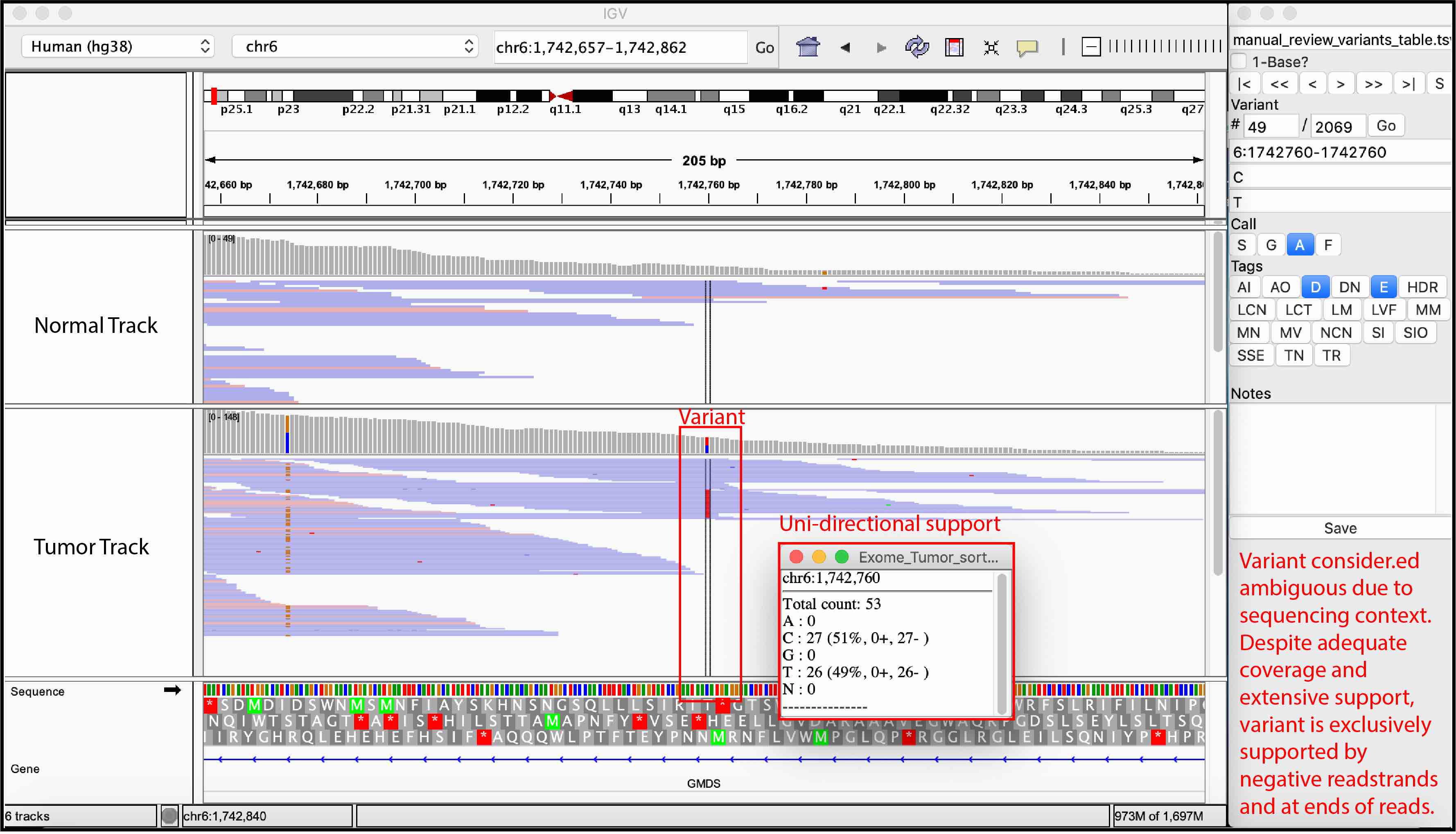
Ambiguous variant
Automated Somatic Variant Refinement
Many of the existing limitations of filtering and manual review could be addressed by automating somatic variant refinement. This would further standardize the MPS pipeline and reduce the labor burden required to identify putative somatic variants. Advancements in computational approaches provide an opportunity for the development of such a process.
To date we have developed DeepSVR, which is a deep learning model that incorporates manual review data from 41,000 variants derived from 9 tumor subtypes in 22 cancer cohorts. This model can be downloaded from the DeepSVR GitHub Repo. A tutorial for DeepSVR can be found on the DeepSVR Wiki.
*Please continue to the next section for instructions on how to perform somatic structural variant calling