Post-Alignment QC
Calculate QC metric for bam files using samtools and picard See docs here: https://github.com/genome/cancer-genomics-workflow/wiki/Alignment
Run Samtools flagstat
cd /workspace/align/
samtools flagstat /workspace/align/Exome_Norm_sorted_mrkdup_bqsr.bam > /workspace/align/Exome_Norm_flagstat.txt
samtools flagstat /workspace/align/Exome_Tumor_sorted_mrkdup_bqsr.bam > /workspace/align/Exome_Tumor_flagstat.txt
# Runtime: < 2min
samtools flagstat /workspace/align/WGS_Norm_merged_sorted_mrkdup_bqsr.bam > /workspace/align/WGS_Norm_merged_flagstat.txt
samtools flagstat /workspace/align/WGS_Tumor_merged_sorted_mrkdup_bqsr.bam > /workspace/align/WGS_Tumor_merged_flagstat.txt
Run various picard CollectMetrics tools
cd /workspace/align/
java -Xmx24g -jar $PICARD CollectInsertSizeMetrics I=/workspace/align/Exome_Norm_sorted_mrkdup_bqsr.bam O=/workspace/align/Exome_Norm_insert_size_metrics.txt H=/workspace/align/Exome_Norm_insert_size_metrics.pdf
java -Xmx24g -jar $PICARD CollectInsertSizeMetrics I=/workspace/align/Exome_Tumor_sorted_mrkdup_bqsr.bam O=/workspace/align/Exome_Tumor_insert_size_metrics.txt H=/workspace/align/Exome_Tumor_insert_size_metrics.pdf
java -Xmx24g -jar $PICARD CollectInsertSizeMetrics I=/workspace/align/WGS_Norm_merged_sorted_mrkdup_bqsr.bam O=/workspace/align/WGS_Norm_merged_insert_size_metrics.txt H=/workspace/align/WGS_Norm_merged_insert_size_metrics.pdf
java -Xmx24g -jar $PICARD CollectInsertSizeMetrics I=/workspace/align/WGS_Tumor_merged_sorted_mrkdup_bqsr.bam O=/workspace/align/WGS_Tumor_merged_insert_size_metrics.txt H=/workspace/align/WGS_Tumor_merged_insert_size_metrics.pdf
# Picard CollectAlignmentSummaryMetrics
java -Xmx24g -jar $PICARD CollectAlignmentSummaryMetrics I=/workspace/align/Exome_Norm_sorted_mrkdup_bqsr.bam O=/workspace/align/Exome_Norm_alignment_metrics.txt R=/workspace/inputs/references/genome/ref_genome.fa
java -Xmx24g -jar $PICARD CollectAlignmentSummaryMetrics I=/workspace/align/Exome_Tumor_sorted_mrkdup_bqsr.bam O=/workspace/align/Exome_Tumor_exome_alignment_metrics.txt R=/workspace/inputs/references/genome/ref_genome.fa
java -Xmx24g -jar $PICARD CollectAlignmentSummaryMetrics I=/workspace/align/WGS_Norm_merged_sorted_mrkdup_bqsr.bam O=/workspace/align/WGS_Norm_merged_alignment_metrics.txt R=/workspace/inputs/references/genome/ref_genome.fa
java -Xmx24g -jar $PICARD CollectAlignmentSummaryMetrics I=/workspace/align/WGS_Tumor_merged_sorted_mrkdup_bqsr.bam O=/workspace/align/WGS_Tumor_merged_alignment_metrics.txt R=/workspace/inputs/references/genome/ref_genome.fa
#Picard CollectHsMetrics
#Exome Only
java -Xmx24g -jar $PICARD CollectHsMetrics I=/workspace/align/Exome_Norm_sorted_mrkdup_bqsr.bam O=/workspace/align/Exome_Norm_hs_metrics.txt R=/workspace/inputs/references/genome/ref_genome.fa BI=/workspace/inputs/references/exome/probe_regions.bed.interval_list TI=/workspace/inputs/references/exome/exome_regions.bed.interval_list
java -Xmx24g -jar $PICARD CollectHsMetrics I=/workspace/align/Exome_Tumor_sorted_mrkdup_bqsr.bam O=/workspace/align/Exome_Tumor_hs_metrics.txt R=/workspace/inputs/references/genome/ref_genome.fa BI=/workspace/inputs/references/exome/probe_regions.bed.interval_list TI=/workspace/inputs/references/exome/exome_regions.bed.interval_list
#Picard CollectGcBiasMetrics
#WGS only
java -Xmx24g -jar $PICARD CollectGcBiasMetrics I=/workspace/align/WGS_Norm_merged_sorted_mrkdup_bqsr.bam O=/workspace/align/WGS_Norm_merged_gc_bias_metrics.txt R=/workspace/inputs/references/genome/ref_genome.fa CHART=/workspace/align/WGS_Norm_merged_gc_bias_metrics.pdf S=/workspace/align/WGS_Norm_merged_gc_bias_summary.txt
java -Xmx24g -jar $PICARD CollectGcBiasMetrics I=/workspace/align/WGS_Tumor_merged_sorted_mrkdup_bqsr.bam O=/workspace/align/WGS_Tumor_merged_gc_bias_metrics.txt R=/workspace/inputs/references/genome/ref_genome.fa CHART=/workspace/align/WGS_Tumor_merged_gc_bias_metrics.pdf S=/workspace/align/WGS_Tumor_merged_gc_bias_summary.txt
#Picard CollectWgsMetrics
#First we need to create the Autosomal Chromosome Interval List
egrep 'chr[0-9]{1,2}\s' /workspace/inputs/references/genome/ref_genome.fa.fai | awk '{print $1"\t1\t"$2"\t+\t"$1}' | cat /workspace/inputs/references/genome/ref_genome.dict - > /workspace/inputs/references/genome/ref_genome_autosomal.interval_list
java -Xmx24g -jar $PICARD CollectWgsMetrics I=/workspace/align/WGS_Norm_merged_sorted_mrkdup_bqsr.bam O=/workspace/align/WGS_Norm_merged_metrics.txt R=/workspace/inputs/references/genome/ref_genome.fa INTERVALS=/workspace/inputs/references/genome/ref_genome_autosomal.interval_list
java -Xmx24g -jar $PICARD CollectWgsMetrics I=/workspace/align/WGS_Tumor_merged_sorted_mrkdup_bqsr.bam O=/workspace/align/WGS_Tumor_merged_metrics.txt R=/workspace/inputs/references/genome/ref_genome.fa INTERVALS=/workspace/inputs/references/genome/ref_genome_autosomal.interval_list
Run FastQC
cd /workspace/align
fastqc -t 8 Exome_Norm_sorted_mrkdup_bqsr.bam
fastqc -t 8 Exome_Tumor_sorted_mrkdup_bqsr.bam
tree
fastqc -t 8 WGS_Norm_merged_sorted_mrkdup_bqsr.bam
fastqc -t 8 WGS_Tumor_merged_sorted_mrkdup_bqsr.bam
tree
#fastqc RNAseq_Norm
#fastqc RNAseq_Tumor
#tree
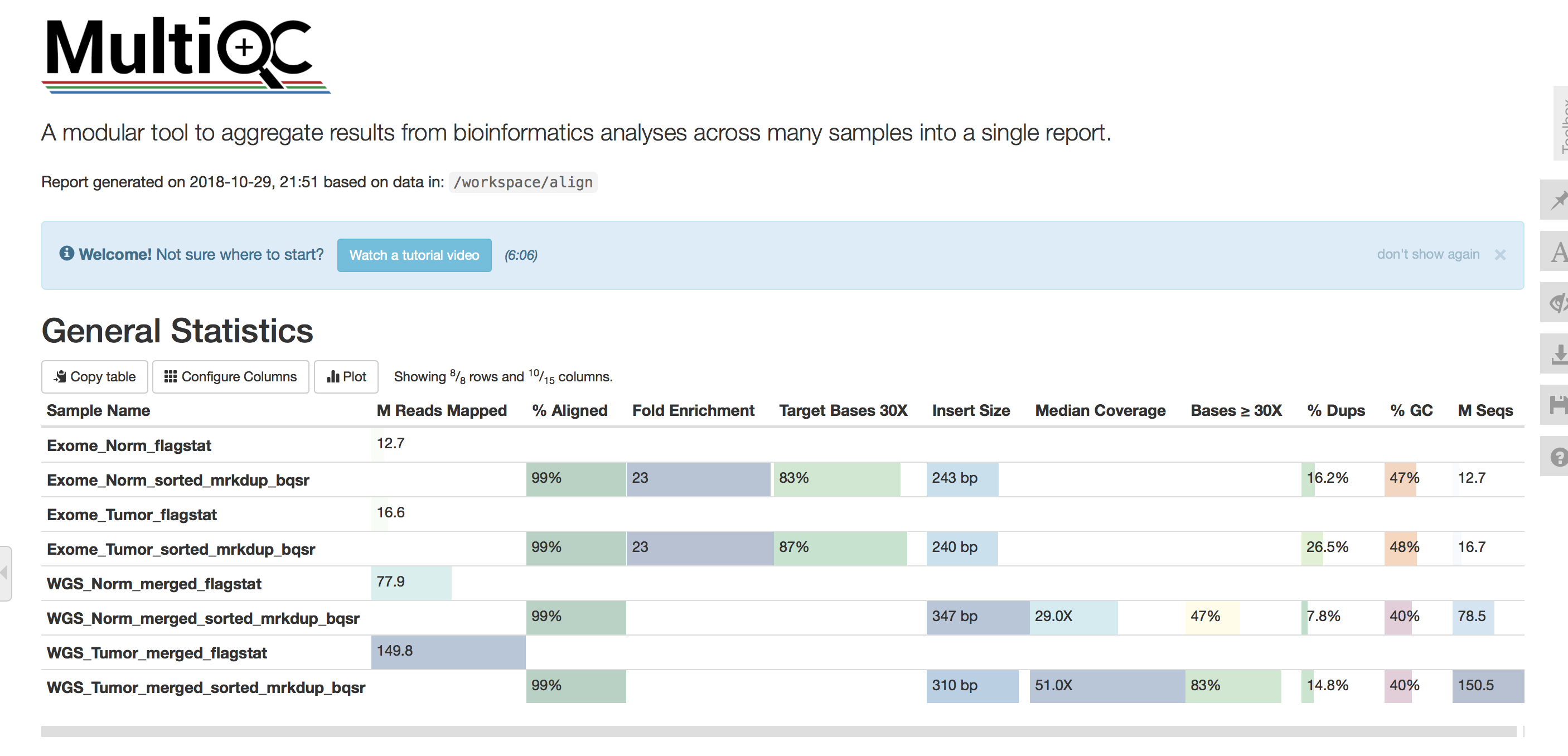
Run MultiQC to produce a final report
cd /workspace/align
mkdir post_align_qc
cd post_align_qc
multiqc /workspace/align/
tree
Here is what the multiqc report should look like, we can view the webpage by loading the html file generated by multiqc: