Germline SNV and Indel Calling
Module objectives
- Perform single-sample germline variant calling with GATK HaplotypeCaller on WGS and exome data
- Perform single-sample germline variant calling with GATK GVCF workflow on WGS and exome data
- Perform single-sample germline variant calling with GATK GVCF workflow on additional exomes from 1000 Genomes Project
- Perform joint genotype calling on exome data, including additional exomes from 1000 Genomes Project
In this module we will use the GATK HaplotypeCaller to call variants from our aligned bams. Since we are only interested in germline variants in this module, we will only call variants in the normal samples (i.e., WGS_Norm and Exome_Norm). The following example commands were inspired by an excellent GATK tutorial, provided by the Broad Institute.
Note: We are using GATK4 v4.0.10.0 for this tutorial.
Run GATK HaplotypeCaller
First, we will run GATK HaplotypeCaller to call germline SNPs and indels. Whenever HaplotypeCaller finds signs of variation it performs a local de novo re-assembly of reads. This improves the accuracy of variant calling, especially in challenging regions, and represents a substantial improvement over the previous GATK UnifiedGenotyper caller. We will also include an option to generate bam output from HaplotypeCaller so that local reassembly/realignment around called variants can be visualized.
GATK HaplotypeCaller is run with the following options:
- –java-options ‘-Xmx60g’ tells GATK to use 60GB of memory
- HaplotypeCaller specifies the GATK command to run
- -R specifies the path to the reference genome
- -I specifies the path to the input bam file for which to call variants
- -O specifies the path to the output vcf file to write variants to
- –bam-output specifies the path to an optional bam file which will store local realignments around variants that HaplotypeCaller used to make calls
- $GATK_REGIONS is an environment variable that we defined earlier to limit calling to specific regions (e.g., just chr6 and chr17)
#Make sure that $GATK_REGIONS is set correctly
echo $GATK_REGIONS
#Create working dir for germline results if not already created
mkdir -p /workspace/germline
cd /workspace/germline
#Call variants for exome data
gatk --java-options '-Xmx60g' HaplotypeCaller -R /workspace/inputs/references/genome/ref_genome.fa -I /workspace/align/Exome_Norm_sorted_mrkdup_bqsr.bam -O /workspace/germline/Exome_Norm_HC_calls.vcf --bam-output /workspace/germline/Exome_Norm_HC_out.bam $GATK_REGIONS
#Call variants for WGS data
gatk --java-options '-Xmx60g' HaplotypeCaller -R /workspace/inputs/references/genome/ref_genome.fa -I /workspace/align/WGS_Norm_merged_sorted_mrkdup_bqsr.bam -O /workspace/germline/WGS_Norm_HC_calls.vcf --bam-output /workspace/germline/WGS_Norm_HC_out.bam $GATK_REGIONS
Exercise - Explore the performance/benefits of local realignment
Let’s examine and compare the alignments made by HaplotypeCaller for local re-alignment around variants to those produced originally by BWA-MEM for the normal (germline) exome data. First start a new IGV session and load the following bam files by URL (don’t forget to substitute your number for #).
- http://s#.pmbio.org/align/Exome_Norm_sorted_mrkdup_bqsr.bam
- http://s#.pmbio.org/germline/Exome_Norm_HC_out.bam
We identified an illustrative deletion by looking through the germline variant calls that we just created in Exome_Norm_HC_calls.vcf. Once you have loaded the above bam files and performed any desired set up (e.g., naming your tracks) navigate to the following coordinates: chr17:76286974-76287203.
You should see something like the following:
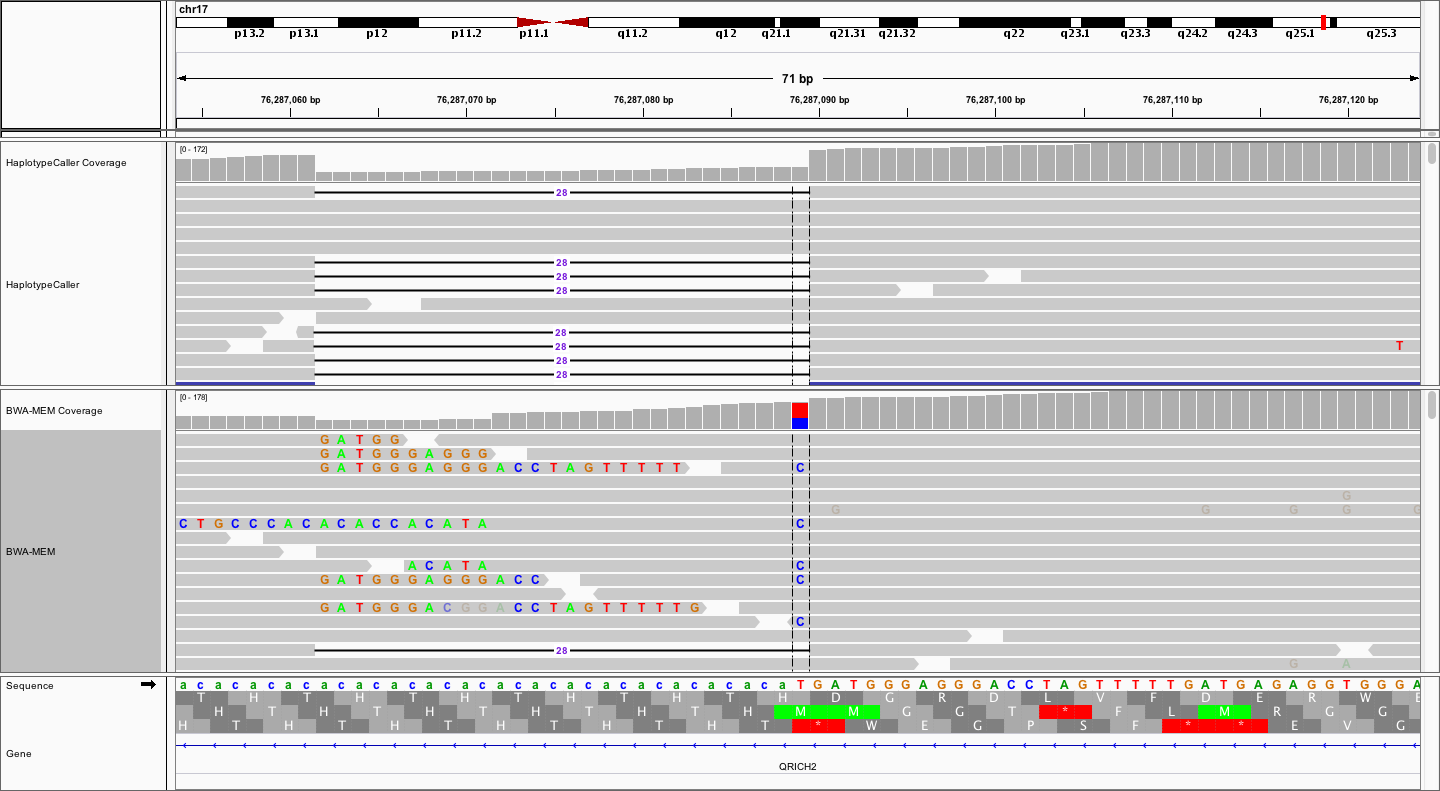
Notice that the local realignment (top track) has produced a very clear call of a 28bp deletion. In contrast, the bwa-mem alignment, while also supporting the 28bp deletion with some reads, is characterized by a bunch of messy soft-clipping and a probably false positive SNV at the end of CA repeat. The contrast is even more striking when we collapse all reads to a squished view.
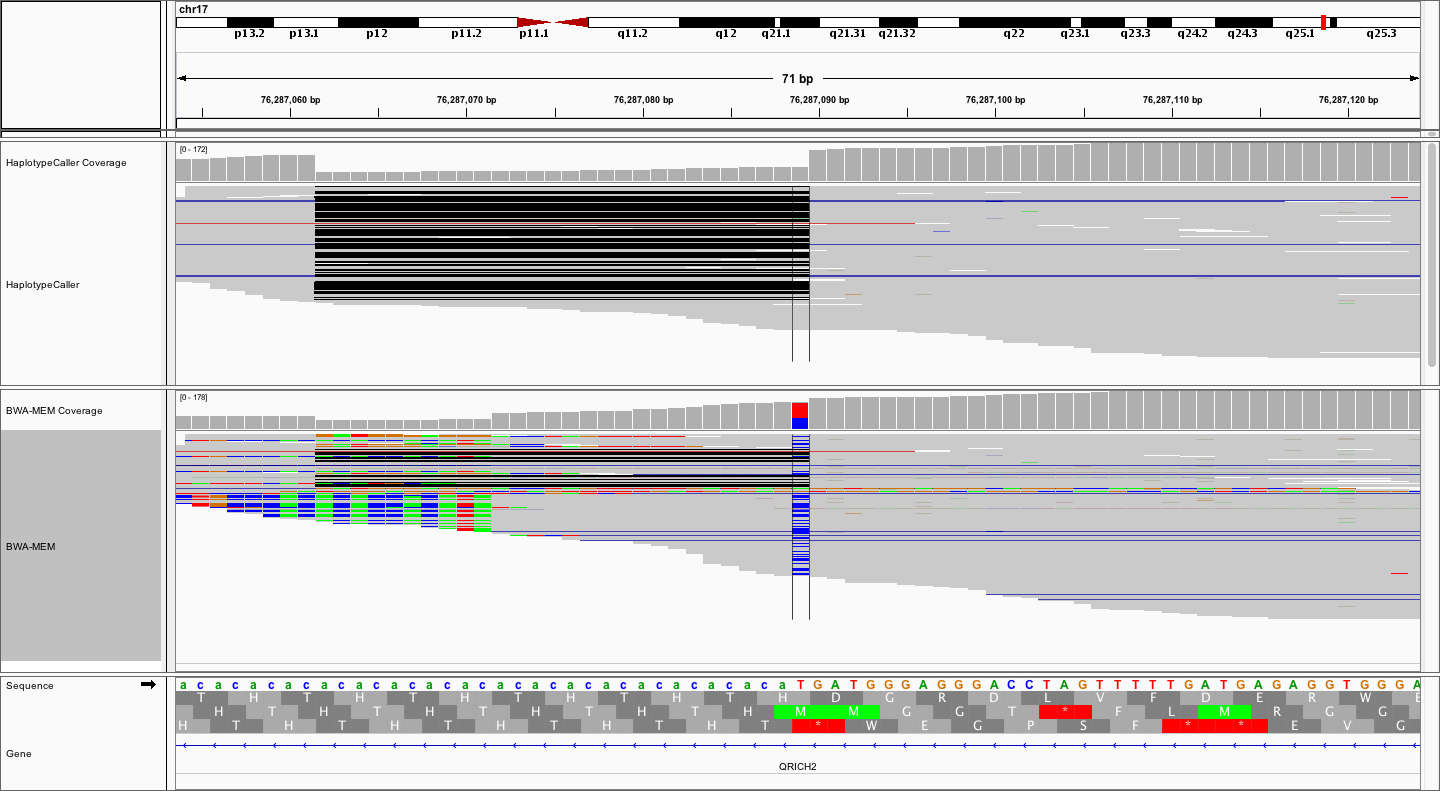
Notice how much “cleaner” the alignments are in the top (locally realigned) track. Unfortunately, many variant callers (without local realignment) would likely call the T->C variant, requiring subsequent filtering and/or manual review to eliminate this artifact. Finally, let’s zoom out a little more to get the big picture.
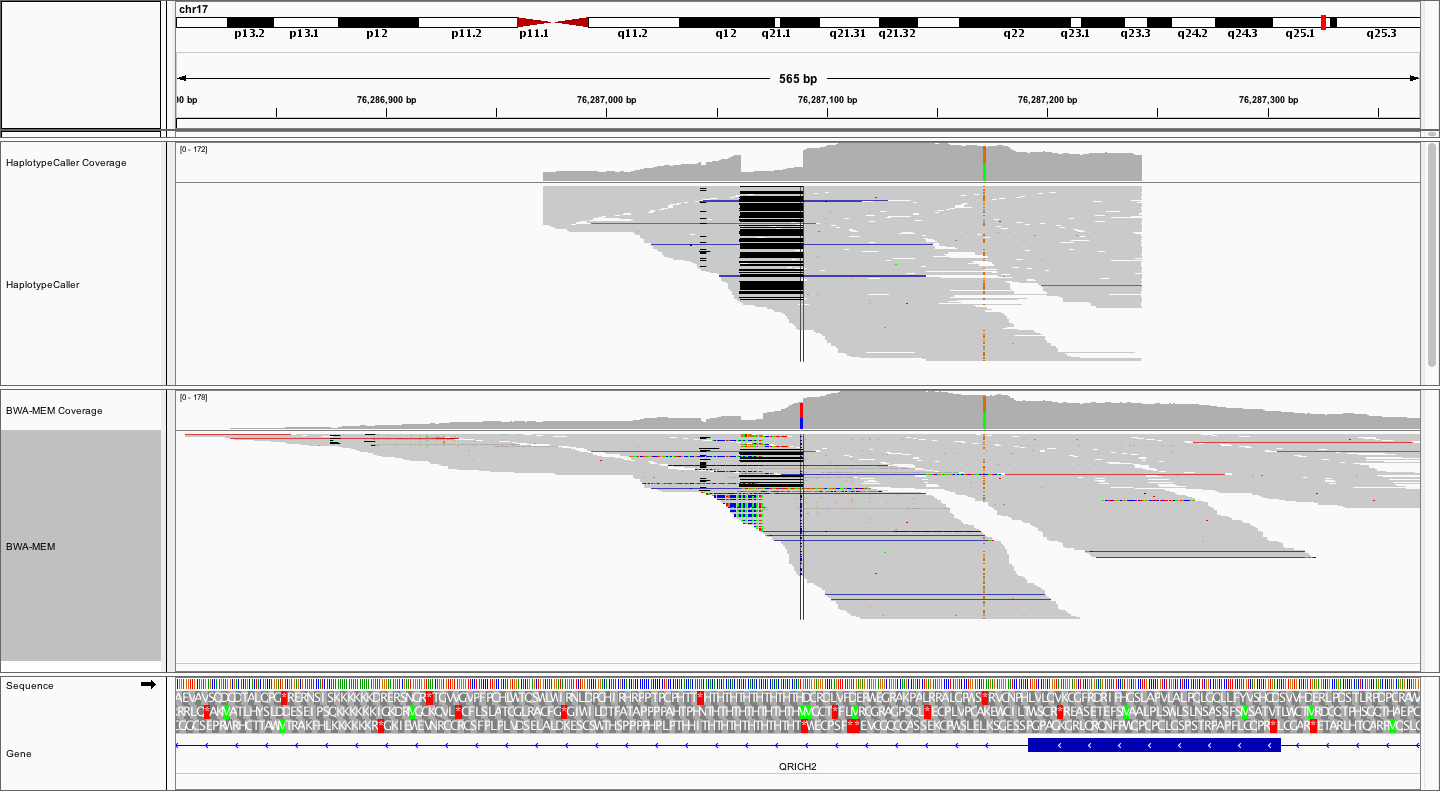
What differences do you notice between the local realignment and bwa-mem alignment from this perspective, apart from the already observed more clean 28bp deletion?
Answer
In the local realignment, the alignments stop abruptly, obviously within some pre-determined window. On the other hand the bwa-mem alignments spread out further, and are naturally distributed around the targeted region (exon). Remember that the local alignment is *local*, not complete. This means that the bam files from HaplotypeCaller represent only a subset of alignments, centered around potential variation. These BAM files are therefore not a replacement for the complete bwa-mem BAMs.
Run HaplotypeCaller in GVCF mode with single sample calling, followed by joint calling (for exomes)
An alternate (and GATK recommended) method is to use the GVCF workflow. In this mode, HaplotypeCaller runs per-sample to generate an intermediate GVCF, which can then be used with the GenotypeGVCF command for joint genotyping of multiple samples in a very efficient way. Joint genotyping has several advantages. In joint genotyping, variants are analyzed across all samples simultaneously. This ensures that if any sample in the study population has variation at a specific site then the genotype for all samples will be determined at that site. It allows more sensitive detection of genotype calls at sites where one sample might have lower coverage but other samples have a confident variant at that position. Joint calling is also necessary to produce data needed for VQSR filtering (see next module). While joint genotyping can be performed directly with HaplotypeCaller (by specifying multiple -I paramters) it is less efficient, and less scalable. Each time a new sample is added the entire (progressively slower) variant calling analysis must be repeated. With the GVCF workflow, new samples can be run individually and then more quickly joint genotyped using the GenotypeGVCFs command.
GATK HaplotypeCaller is run with all of the same options as above, except for one addition:
- -ERC GVCF specifies to use the GCVF workflow, producing a GVCF instead of VCF
#Call variants in GVCF mode for exome data
gatk --java-options '-Xmx60g' HaplotypeCaller -ERC GVCF -R /workspace/inputs/references/genome/ref_genome.fa -I /workspace/align/Exome_Norm_sorted_mrkdup_bqsr.bam -O /workspace/germline/Exome_Norm_HC_calls.g.vcf --bam-output /workspace/germline/Exome_Norm_HC_GVCF_out.bam $GATK_REGIONS
#Call variants in GVCF mode for WGS data - currently this takes too long and isn't used for any subsequent modules. Skip for now.
#gatk --java-options '-Xmx60g' HaplotypeCaller -ERC GVCF -R /workspace/inputs/references/genome/ref_genome.fa -I /workspace/align/WGS_Norm_merged_sorted_mrkdup_bqsr.bam -O /workspace/germline/WGS_Norm_HC_calls.g.vcf --bam-output /workspace/germline/WGS_Norm_HC_GVCF_out.bam $GATK_REGIONS
Future improvement: Experiment with splitting the above GVCF commands into smaller segments for faster run times.
Obtain 1000 Genomes Project (1KGP) exome data for joint genotyping and VQSR steps
As described above, there are several advantages to joint genotype calling. The additional samples and their variants improve the overall accuracy of variant calling and subsequent filtering steps. However, in this case we only have one “normal” sample, our HCC1395BL cell line. Therefore, we will make use of a set of public exome sequencing data to illustrate multi-sample joint calling. The 1000 Genome Project is a large international effort to create a set of publicly available sequence data for a large cohort of ethnically diverse individuals. Recall that the cell line being used in this tutorial (HCC1395) was derived from a 43 year old caucasian female (by a research group in Dallas Texas). Therefore, for a “matched” set of reference alignments we might limit to those of European descent.
Explore the available 1000 Genome samples
- First, view a summary of samples/ethnicities available from the 1000 Genomes Populations page
- Now, we are guessing, but perhaps GBR (British in England and Scotland) would be the best match.
- Go to the 1000 Genomes Samples page
- Filter by population -> GBR
- Filter by analysis group -> Exome
- Filter by data collection -> 1000 genomes on GRCh38
- Download the list to get sample details (e.g., save as igsr_samples_GBR.tsv)
Get a list of data files for GBR samples with exome data
- Go to back to the 1000 Genomes Populations page
- Select GBR
- Scroll down to ‘Data collections for GBR’
- Choose ‘1000 Genomes on GRCh38’ tab
- Select ‘Data types’ -> ‘Alignment’
- Select ‘Analysis groups’ -> ‘Exome’
- ‘Download the list’ (e.g., save as igsr_GBR_GRCh38_exome_alignment.tsv).
Using the information obtained above, we could download the already aligned exome data for several 1KGP individuals in cram format. These cram files were created in a generally compatible way with the anlysis done so far in this tutorial. They were aligned with BWA-mem to GRCh38 with alternative sequences, plus decoys and HLA. This was followed by GATK BAM improvement steps as in the 1000 Genomes phase 3 pipeline (GATK IndelRealigner, BQSR, and Picard MarkDuplicates). Of course, there will be batch effects related to potentially different sample preparation, library construction, exome capture reagent and protocol, sequencing pipeline, etc. For more details see:
- http://www.internationalgenome.org/analysis
- https://media.nature.com/original/nature-assets/nature/journal/v526/n7571/extref/nature15393-s1.pdf
For expediency, in preparation for this course, we downloaded exome alignments for 5 females, of GBR descent. We limited that data to just chr6 and/or chr17 and then performed germline variant calling using the GATK HaplotypeCaller GVCF workflow commands as above. The details for this can be found on the Developer’s Notes page.
Download pre-computed GVCFs and their index files for five 1KGP exomes that we will use for joint genotyping.
cd /workspace/germline/
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00099_HC_calls.g.vcf
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00099_HC_calls.g.vcf.idx
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00102_HC_calls.g.vcf
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00102_HC_calls.g.vcf.idx
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00104_HC_calls.g.vcf
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00104_HC_calls.g.vcf.idx
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00150_HC_calls.g.vcf
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00150_HC_calls.g.vcf.idx
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00158_HC_calls.g.vcf
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00158_HC_calls.g.vcf.idx
Perform joint genotype calling
Create a joint genotype GVCF, combining HCC1395 Exome normal together with a set of 1KGP exomes, for later use in VQSR filtering.
First, combine the g.vcf files together into a single combined g.vcf with the CombineGVCFs command.
GATK CombineGVCFs is run with the following options:
- –java-options ‘-Xmx60g’ tells GATK to use 60GB of memory
- CombineGVCFs specifies the GATK command to run
- -R specifies the path to the reference genome
- -V [multiple] specifies the path to each of the input g.vcf files
- -O specifies the path to the output combined g.vcf file
- $GATK_REGIONS is an environment variable that we defined earlier to limit calling to specific regions (e.g., just chr6 and chr17)
gatk --java-options '-Xmx60g' CombineGVCFs -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.g.vcf -V /workspace/germline/HG00099_HC_calls.g.vcf -V /workspace/germline/HG00102_HC_calls.g.vcf -V /workspace/germline/HG00104_HC_calls.g.vcf -V /workspace/germline/HG00150_HC_calls.g.vcf -V /workspace/germline/HG00158_HC_calls.g.vcf -O /workspace/germline/Exome_Norm_1KGP_HC_calls_combined.g.vcf $GATK_REGIONS
Finally, perfom joint genotyping with the GenotypeGVCFs command. This command is run with the following options:
- –java-options ‘-Xmx60g’ tells GATK to use 60GB of memory
- GenotypeGVCFs specifies the GATK command to run
- -R specifies the path to the reference genome
- -V specifies the path to combined g.vcf files
- -O specifies the path to the output the joint genotyped vcf file
- $GATK_REGIONS is an environment variable that we defined earlier to limit calling to specific regions (e.g., just chr6 and chr17)
gatk --java-options '-Xmx60g' GenotypeGVCFs -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_1KGP_HC_calls_combined.g.vcf -O /workspace/germline/Exome_GGVCFs_jointcalls.vcf $GATK_REGIONS
Acknowledgements and citations
This analysis demonstrated in this tutorial would not be possible without the efforts of developers who wrote and maintain the GATK suite of tools. We have also acknowledged their excellent tutorials, documentation, forums, and workshop materials wherever possible. Similarly, this tutorial benefits from the public availability of data from the 1000 Genomes Project. The GATK and 1000 Genomes papers are also cited below. Please remember to cite these tools in your publications when using them.
- The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. Genome Res. 2010 Sep;20(9):1297-303. doi: 10.1101/gr.107524.110. Epub 2010 Jul 19.
- A framework for variation discovery and genotyping using next-generation DNA sequencing data. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. Nat Genet. 2011 May;43(5):491-8. doi: 10.1038/ng.806. Epub 2011 Apr 10.
- From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. Curr Protoc Bioinformatics. 2013;43:11.10.1-33. doi: 10.1002/0471250953.bi1110s43.
- A global reference for human genetic variation. 1000 Genomes Project Consortium. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. Nature. 2015 Oct 1;526(7571):68-74. doi: 10.1038/nature15393.
Future developments
- Consider implementing the following options: “–max_alternate_alleles 3 -variant_index_type LINEAR -variant_index_parameter 128000 -contamination $freemix –read_filter OverclippedRead”. See: https://confluence.ris.wustl.edu/display/BIO/Proposed+CCDG+Analysis+Workflow+-+2017.7.14
- Consider demonstrating GenomicsDBImport as an alternative to CombineGVCFs for when larger numbers of samples are to merged before GenotypeGVCFs