Germline SNV/Indel Filtering/Annotation/Review
Module objectives
- Perform GATK hard-filtering of germline SNVs and indels
- Perform GATK VQSR-filtering of germline SNVs and indels
- Perform VEP annotation of filtered variants.
In this module we will learn about variant filtering and annotation. The raw output of GATK HaplotypeCaller will include many variants with varying degrees of quality. For various reasons we might wish to further filter these to a higher confidence set of variants. The recommended approach is to use GATK VQSR (see below for more details). This requires a large number of variants. Sufficient numbers may be available for even a single sample of whole genome sequencing data. However for targeted sequencing (e.g., exome data) it is recommended to include a larger number (i.e., at least 30-50), preferably platform-matched (i.e., similar sequencing strategy), samples with variant calls. For our purposes, we will first demonstrate a less optimal hard-filtering strategy. Then we will demonstrate VQSR filtering.
It is strongly recommended to read the following documentation from GATK:
Perform hard-filtering on Exome data
Extract SNPs and Indels from the variant call set
First, we will separate out the SNPs and Indels from the VCF into new separate VCFs. Note that the variant type (SNP, INDEL, MIXED, etc) is not stored explicitly in the vcf but instead inferred from the genotypes. We will use a versatile GATK tool called SelectVariants. This command can be used for all kinds of simple filtering or subsetting purposes. We will run it twice to select by variant type, once for SNPs, and then again for Indels, to produce two new VCFs.
GATK SelectVariants is run with the following options:
- –java-options ‘-Xmx60g’ tells GATK to use 60GB of memory
- SelectVariants specifies the GATK command to run
- -R specifies the path to the reference genome
- -V specifies the path to the input vcf file to be filtered
- -select-type [SNP, INDEL, MIXED, etc] specifies which type of variant to limit to
- -O specifies the path to the output vcf file to be produced
cd /workspace/germline/
gatk --java-options '-Xmx60g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.vcf -select-type SNP -O /workspace/germline/Exome_Norm_HC_calls.snps.vcf
gatk --java-options '-Xmx60g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.vcf -select-type INDEL -O /workspace/germline/Exome_Norm_HC_calls.indels.vcf
Apply basic filters to the SNP and Indel call sets
Next, we will perform so-called hard-filtering by applying a number of hard (somewhat arbitrary) cutoffs. For example, we might require each variant to have a minimum QualByDepth (QD) of 2.0. The QD value is the variant confidence (from the QUAL field) divided by the unfiltered depth of non-reference samples. With such a filter any variant with a QD value less than 2.0 would be marked as filtered in the FILTER field with a filter name of our choosing (e.g., QD_lt_2). Multiple filters can be combined arbitrarily. Each can be given its own name so that you can later determine which one or more filters a variant fails. Visit the GATK documentation on hard-filtering to learn more about the following hard filtering options. Notice that different filters and cutoffs are recommended for SNVs and Indels. This is why we first split them into separate files.
cd /workspace/germline/
gatk --java-options '-Xmx64g' VariantFiltration -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.snps.vcf --filter-expression "QD < 2.0" --filter-name "QD_lt_2" --filter-expression "FS > 60.0" --filter-name "FS_gt_60" --filter-expression "MQ < 40.0" --filter-name "MQ_lt_40" --filter-expression "MQRankSum < -12.5" --filter-name "MQRS_lt_n12.5" --filter-expression "ReadPosRankSum < -8.0" --filter-name "RPRS_lt_n8" --filter-expression "SOR > 3.0" --filter-name "SOR_gt_3" -O /workspace/germline/Exome_Norm_HC_calls.snps.filtered.vcf
gatk --java-options '-Xmx64g' VariantFiltration -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.indels.vcf --filter-expression "QD < 2.0" --filter-name "QD_lt_2" --filter-expression "FS > 200.0" --filter-name "FS_gt_200" --filter-expression "ReadPosRankSum < -20.0" --filter-name "RPRS_lt_n20" --filter-expression "SOR > 10.0" --filter-name "SOR_gt_10" -O /workspace/germline/Exome_Norm_HC_calls.indels.filtered.vcf
Let’s take a look at a few of the variants in one of these filtered files. Remember, we haven’t actually removed any variants. We have just marked them as PASS or filter-name. Use grep -v and head to skip past all the VCF header lines and view the first few records. How many variants passed or failed our filters?
grep -v "##" Exome_Norm_HC_calls.snps.filtered.vcf | head -10
Merge filtered SNP and INDEL vcfs back together
It is convenient to have all our variants in a single result file. Therfore, we will merge them back together.
GATK MergeVcfs is run with the following options:
- –java-options ‘-Xmx60g’ tells GATK to use 60GB of memory
- MergeVcfs specifies the GATK command to run
- -I specifies the path to each of the vcf files to be merged
- -O specifies the path to the output vcf file to be produced
gatk --java-options '-Xmx60g' MergeVcfs -I /workspace/germline/Exome_Norm_HC_calls.snps.filtered.vcf -I /workspace/germline/Exome_Norm_HC_calls.indels.filtered.vcf -O /workspace/germline/Exome_Norm_HC_calls.filtered.vcf
Extract PASS variants only
It would also be convenient to have a vcf with only passing variants. For this, we can go back the GATK SelectVariants tool. This will be run much as above except with the --exlude-filtered option instead of -select-type.
GATK SelectVariants is run with the following options:
- –java-options ‘-Xmx60g’ tells GATK to use 60GB of memory
- SelectVariants specifies the GATK command to run
- -R specifies the path to the reference genome
- -V specifies the path to the input vcf file to be filtered
- –exclude-filtered specifies to remove all variants except those marked as PASS
- -O specifies the path to the output vcf file to be produced
gatk --java-options '-Xmx60g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.filtered.vcf -O /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vcf --exclude-filtered
Perform hard-filtering on WGS data
Hard-filtering on the WGS dataset will be performed nearly identically to above.
# Extract the SNPs from the call set
gatk --java-options '-Xmx60g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/WGS_Norm_HC_calls.vcf -select-type SNP -O /workspace/germline/WGS_Norm_HC_calls.snps.vcf
# Extract the indels from the call set
gatk --java-options '-Xmx64g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/WGS_Norm_HC_calls.vcf -select-type INDEL -O /workspace/germline/WGS_Norm_HC_calls.indels.vcf
# Apply basic filters to the SNP call set
gatk --java-options '-Xmx64g' VariantFiltration -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/WGS_Norm_HC_calls.snps.vcf --filter-expression "QD < 2.0" --filter-name "QD_lt_2" --filter-expression "FS > 60.0" --filter-name "FS_gt_60" --filter-expression "MQ < 40.0" --filter-name "MQ_lt_40" --filter-expression "MQRankSum < -12.5" --filter-name "MQRS_lt_n12.5" --filter-expression "ReadPosRankSum < -8.0" --filter-name "RPRS_lt_n8" -O /workspace/germline/WGS_Norm_HC_calls.snps.filtered.vcf
# Apply basic filters to the INDEL call set
gatk --java-options '-Xmx64g' VariantFiltration -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/WGS_Norm_HC_calls.indels.vcf --filter-expression "QD < 2.0" --filter-name "QD_lt_2" --filter-expression "FS > 200.0" --filter-name "FS_gt_200" --filter-expression "ReadPosRankSum < -20.0" --filter-name "RPRS_lt_n20" -O /workspace/germline/WGS_Norm_HC_calls.indels.filtered.vcf
# Merge filtered SNP and INDEL vcfs back together
gatk --java-options '-Xmx64g' MergeVcfs -I /workspace/germline/WGS_Norm_HC_calls.snps.filtered.vcf -I /workspace/germline/WGS_Norm_HC_calls.indels.filtered.vcf -O /workspace/germline/WGS_Norm_HC_calls.filtered.vcf
# Extract PASS variants only
gatk --java-options '-Xmx64g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/WGS_Norm_HC_calls.filtered.vcf -O /workspace/germline/WGS_Norm_HC_calls.filtered.PASS.vcf --exclude-filtered
NOTE: There are a number of MIXED type variants (multi-allelic with both SNP and INDEL alleles) that are currently dropped by the above workflow (selecting for only SNPs and INDELs). In the future we could consider converting these with gatk LeftAlignAndTrimVariants –split-multi-allelics.
Perform VQSR-filtering of Exome variants that were joint called with 1KG exomes
VQSR filtering is a much more sophisticated approach than hard-filtering. In this approach a number of reference variant call sets are provided as truth/training data from which to build a model. This model estimates the probability that a variant is real and allows filtering at various confidence levels.
When using VQSR filtering on exome data, there will be a smaller number of variants per sample compared to WGS. These are typically insufficient to build a robust recalibration model. If running on only a few samples, GATK recommends that you analyze samples jointly in cohorts of at least 30 samples. If necessary, add exomes from 1000 Genome Project (1KGP) or comparable. Ideally, these should be processed with similar technical generation (technology, capture, read length, depth). Recall that we performed joint genotyping on our exome normal sample along with 5 1KGP exomes to illustrate this concept.
The VQSR process will be briefly demonstrated below. Visit the GATK documentation on VQSR-filtering to learn more about how to correctly perform Variant recalibration with VQSR. See also: https://drive.google.com/drive/folders/0BzI1CyccGsZiWlU5SXNvNnFGbVE (GATK Workshops -> 1709 -> GATKwr19-05-Variant_filtering.pdf)
Build the SNP recalibration model
First, we will build a model to predict high confidence SNPs using various known SNP datasets and their intrinsic features.
GATK VariantRecalibrator is run with the following options:
- –java-options ‘-Xmx60g’ tells GATK to use 60GB of memory
- VariantRecalibrator specifies the GATK command to run
- -R specifies the path to the reference genome
- -V specifies the path to the input vcf file to be filtered
- –resource [multiple] specific variant call sets to be used for training
- -an specifies the variant features to use for modeling
- –mode specifies SNP or INDEL mode
- -tranche specifies difference confidence levels to mark variants with
- -O specifies the path for the recalibration file to be produced
- –tranches-file specifies the path for the tranches data to be saved
-
–rscript-file specifies the path for the R script to be saved
- Note: Any annotations specified (“-an XX”) below must actually be present in your VCF. They should have been added at an earlier step in the GATK workflow. If not, they could be added with VariantAnnotator
- Note: For exome data, exclude “-an DP” as this coverage metric should only be used if extreme deviations in coverage are not expected and indicative of errors
- Note: The “-an InbreedingCoeff” option is for a population level statistic that requires at least 10 samples in order to be computed (When?). For projects with fewer samples, or that includes many closely related samples (such as a family) omit this annotation from the command line.
cd /workspace/germline/
gatk --java-options '-Xmx60g' VariantRecalibrator -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_GGVCFs_jointcalls.vcf --resource hapmap,known=false,training=true,truth=true,prior=15.0:/workspace/inputs/references/gatk/hapmap_3.3.hg38.vcf.gz --resource omni,known=false,training=true,truth=true,prior=12.0:/workspace/inputs/references/gatk/1000G_omni2.5.hg38.vcf.gz --resource 1000G,known=false,training=true,truth=false,prior=10.0:/workspace/inputs/references/gatk/1000G_phase1.snps.high_confidence.hg38.vcf.gz --resource dbsnp,known=true,training=false,truth=false,prior=2.0:/workspace/inputs/references/gatk/Homo_sapiens_assembly38.dbsnp138.vcf.gz -an QD -an FS -an SOR -an MQ -an MQRankSum -an ReadPosRankSum --mode SNP -tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 -O recalibrate_SNP.recal --tranches-file recalibrate_SNP.tranches --rscript-file recalibrate_SNP_plots.R
Apply recalibration to SNPs
Next, we will apply this model to our called SNPs.
GATK ApplyVQSR is run with the following options:
- –java-options ‘-Xmx60g’ tells GATK to use 60GB of memory
- ApplyVQSR specifies the GATK command to run
- -R specifies the path to the reference genome
- -V specifies the path to the input vcf file to be filtered
- –mode specifies SNP or INDEL mode
- –truth-sensitivity-filter-level specifies the confidence level to apply
- –recal-file specifies the path to recalibrarion file created above
- –tranches-file specifies the path to tranches file created above
- -O specifies the path for the recalibrated variant file to be output to
gatk --java-options '-Xmx60g' ApplyVQSR -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_GGVCFs_jointcalls.vcf --mode SNP --truth-sensitivity-filter-level 99.0 --recal-file /workspace/germline/recalibrate_SNP.recal --tranches-file /workspace/germline/recalibrate_SNP.tranches -O /workspace/germline/Exome_GGVCFs_jointcalls_recalibrated_snps_raw_indels.vcf
Build and apply Indel recalibration
A model for filtering indels is built and applied in much the same manner as for SNPs above.
#Build Indel recalibration model
gatk --java-options '-Xmx64g' VariantRecalibrator -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_GGVCFs_jointcalls_recalibrated_snps_raw_indels.vcf --resource mills,known=false,training=true,truth=true,prior=12.0:/workspace/inputs/references/gatk/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz --resource dbsnp,known=true,training=false,truth=false,prior=2.0:/workspace/inputs/references/gatk/Homo_sapiens_assembly38.dbsnp138.vcf.gz -an QD -an FS -an SOR -an MQRankSum -an ReadPosRankSum --mode INDEL -tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 --max-gaussians 4 -O recalibrate_INDEL.recal --tranches-file recalibrate_INDEL.tranches --rscript-file recalibrate_INDEL_plots.R
#Apply recalibration to Indels
gatk --java-options '-Xmx64g' ApplyVQSR -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_GGVCFs_jointcalls_recalibrated_snps_raw_indels.vcf --mode INDEL --truth-sensitivity-filter-level 99.0 --recal-file /workspace/germline/recalibrate_INDEL.recal --tranches-file /workspace/germline/recalibrate_INDEL.tranches -O /workspace/germline/Exome_GGVCFs_jointcalls_recalibrated.vcf
Extract PASS variants only and only variants actually called and non-reference in our sample of interest
Once again, we will use GATK SelectVariants to create a new vcf of just variants with FILTER = PASS.
gatk --java-options '-Xmx64g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_GGVCFs_jointcalls_recalibrated.vcf -O /workspace/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vcf --exclude-filtered --exclude-non-variants --remove-unused-alternates --sample-name HCC1395BL_DNA
Perform VQSR-filtering of WGS variants
As above, we can apply VQSR filtering to the WGS data.
cd /workspace/germline/
#Build SNP recalibration model
gatk --java-options '-Xmx64g' VariantRecalibrator -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/WGS_Norm_HC_calls.vcf --resource hapmap,known=false,training=true,truth=true,prior=15.0:/workspace/inputs/references/gatk/hapmap_3.3.hg38.vcf.gz --resource omni,known=false,training=true,truth=true,prior=12.0:/workspace/inputs/references/gatk/1000G_omni2.5.hg38.vcf.gz --resource 1000G,known=false,training=true,truth=false,prior=10.0:/workspace/inputs/references/gatk/1000G_phase1.snps.high_confidence.hg38.vcf.gz --resource dbsnp,known=true,training=false,truth=false,prior=2.0:/workspace/inputs/references/gatk/Homo_sapiens_assembly38.dbsnp138.vcf.gz -an QD -an FS -an SOR -an MQ -an MQRankSum -an ReadPosRankSum -an DP --mode SNP -tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 -O recalibrate_SNP.WGS.recal --tranches-file recalibrate_SNP.WGS.tranches --rscript-file recalibrate_SNP_plots.WGS.R
#Apply recalibration to SNPs
gatk --java-options '-Xmx64g' ApplyVQSR -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/WGS_Norm_HC_calls.vcf --mode SNP --truth-sensitivity-filter-level 99.0 --recal-file /workspace/germline/recalibrate_SNP.WGS.recal --tranches-file /workspace/germline/recalibrate_SNP.WGS.tranches -O /workspace/germline/WGS_Norm_HC_calls_recalibrated_snps_raw_indels.vcf
#Build Indel recalibration model
gatk --java-options '-Xmx64g' VariantRecalibrator -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/WGS_Norm_HC_calls_recalibrated_snps_raw_indels.vcf --resource mills,known=false,training=true,truth=true,prior=12.0:/workspace/inputs/references/gatk/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz --resource dbsnp,known=true,training=false,truth=false,prior=2.0:/workspace/inputs/references/gatk/Homo_sapiens_assembly38.dbsnp138.vcf.gz -an QD -an FS -an SOR -an MQRankSum -an ReadPosRankSum -an DP --mode INDEL -tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 --max-gaussians 4 -O recalibrate_INDEL.WGS.recal --tranches-file recalibrate_INDEL.WGS.tranches --rscript-file recalibrate_INDEL_plots.WGS.R
#Apply recalibration to Indels
gatk --java-options '-Xmx64g' ApplyVQSR -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/WGS_Norm_HC_calls_recalibrated_snps_raw_indels.vcf --mode INDEL --truth-sensitivity-filter-level 99.0 --recal-file /workspace/germline/recalibrate_INDEL.WGS.recal --tranches-file /workspace/germline/recalibrate_INDEL.WGS.tranches -O /workspace/germline/WGS_Norm_HC_calls_recalibrated.vcf
#Extract PASS variants only and only variants actually called and non-reference in our sample of interest
gatk --java-options '-Xmx64g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/WGS_Norm_HC_calls_recalibrated.vcf -O /workspace/germline/WGS_Norm_HC_calls_recalibrated.PASS.vcf --exclude-filtered --exclude-non-variants --remove-unused-alternates --sample-name HCC1395BL_DNA
Exercise
Choose one of the filtering strategies above, try changing the filter criteria to increase or decrease the stringency of various filters, and then view the result effect on the numbers of variants passing filters.
Perform VEP annotation of filtered variants
Now that we have high-confidence, filtered variants, we want to start understanding which of these variants might be clinically or biologically relevant. Ensembl’s VEP annotation software is a powerful tool for annotating variants with a great deal of biological features. This includes such information as protein consequence (non-coding or coding), population frequencies, links to external databases, various scores designed to estimate the importance of individual variants on protein function, and much more.
VEP is run with the following options:
- –cache enables use of the local cache with limited network access
- –dir_cache specifies the cache directory to use
- –dir_plugins specifies the plugin directory to use
- –fasta specifies a FASTA file to use for reference genome sequence
- –fork specifies number of cpus to fork job across
- –assembly specifies the assembly version to use
- –offline runs in offline mode
- –vcf/–tab specifies to output annotated results in VCF or tab-delimited format
- –plugin [multiple] specifies to use the named plugin. In this case the Downstream plugin is specified to add downstream amino acid consequences of frameshift mutations
- –everything is a shortcut flag to turn on a number of other options (–sift b, –polyphen b, –ccds, etc). See VEP docs.
- –terms specify the type of consequence terms to output. In this case we chose SO - Sequence Ontology.
- –pick pick one line or block of consequence data per variant, including transcript-specific column
- –coding_only specifies to only return consequences that fall in the coding regions of transcripts
- –transcript_version specifies to add version numbers to Ensembl transcript identifiers
- -i specifies path to input VCF file to be annotated
- -o specifies path to output VCF/TSV file to be produced
-
–force_overwrite specifies to force the overwrite of an existing file without warning
- Visit the VEP documentation for more details on how to run VEP.
- Note, the first time you run VEP it will create a fasta index for the reference genome in VEP cache. Therefore, it will take longer to run that first time but should speed up substantially for subsequent runs on files with similar numbers of variants.
We will run VEP several times to annotate hard-filtered exome/WGS and VQSR-filter exome/WGS each with both VCF and TSV output for a total of 8 VEP runs
#VEP annotate hard-filtered exome results
#Output VEP VCF
vep --cache --dir_cache /opt/vep_cache --dir_plugins /opt/vep_cache/Plugins --fasta /opt/vep_cache/homo_sapiens/93_GRCh38/Homo_sapiens.GRCh38.dna.toplevel.fa.gz --fork 8 --assembly=GRCh38 --offline --vcf --plugin Downstream --everything --terms SO --pick --coding_only --transcript_version -i /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vcf -o /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vep.vcf --force_overwrite
#Output tabular VEP
vep --cache --dir_cache /opt/vep_cache --dir_plugins /opt/vep_cache/Plugins --fasta /opt/vep_cache/homo_sapiens/93_GRCh38/Homo_sapiens.GRCh38.dna.toplevel.fa.gz --fork 8 --assembly=GRCh38 --offline --tab --plugin Downstream --everything --terms SO --pick --coding_only --transcript_version -i /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vcf -o /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vep.tsv --force_overwrite
#VEP annotate hard-filtered WGS results
#Output VEP VCF
vep --cache --dir_cache /opt/vep_cache --dir_plugins /opt/vep_cache/Plugins --fasta /opt/vep_cache/homo_sapiens/93_GRCh38/Homo_sapiens.GRCh38.dna.toplevel.fa.gz --fork 8 --assembly=GRCh38 --offline --vcf --plugin Downstream --everything --terms SO --pick --coding_only --transcript_version -i /workspace/germline/WGS_Norm_HC_calls.filtered.PASS.vcf -o /workspace/germline/WGS_Norm_HC_calls.filtered.PASS.vep.vcf --force_overwrite
#Output tabular VEP
vep --cache --dir_cache /opt/vep_cache --dir_plugins /opt/vep_cache/Plugins --fasta /opt/vep_cache/homo_sapiens/93_GRCh38/Homo_sapiens.GRCh38.dna.toplevel.fa.gz --fork 8 --assembly=GRCh38 --offline --tab --plugin Downstream --everything --terms SO --pick --coding_only --transcript_version -i /workspace/germline/WGS_Norm_HC_calls.filtered.PASS.vcf -o /workspace/germline/WGS_Norm_HC_calls.filtered.PASS.vep.tsv --force_overwrite
#VEP annotate VQSR-filtered exome results
#Output VEP VCF
vep --cache --dir_cache /opt/vep_cache --dir_plugins /opt/vep_cache/Plugins --fasta /opt/vep_cache/homo_sapiens/93_GRCh38/Homo_sapiens.GRCh38.dna.toplevel.fa.gz --fork 8 --assembly=GRCh38 --offline --vcf --plugin Downstream --everything --terms SO --pick --coding_only --transcript_version -i /workspace/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vcf -o /workspace/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vep.vcf --force_overwrite
#Output tabular VEP
vep --cache --dir_cache /opt/vep_cache --dir_plugins /opt/vep_cache/Plugins --fasta /opt/vep_cache/homo_sapiens/93_GRCh38/Homo_sapiens.GRCh38.dna.toplevel.fa.gz --fork 8 --assembly=GRCh38 --offline --tab --plugin Downstream --everything --terms SO --pick --coding_only --transcript_version -i /workspace/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vcf -o /workspace/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vep.tsv --force_overwrite
#VEP annotate VQSR-filtered WGS results
#Output VEP VCF
vep --cache --dir_cache /opt/vep_cache --dir_plugins /opt/vep_cache/Plugins --fasta /opt/vep_cache/homo_sapiens/93_GRCh38/Homo_sapiens.GRCh38.dna.toplevel.fa.gz --fork 8 --assembly=GRCh38 --offline --vcf --plugin Downstream --everything --terms SO --pick --coding_only --transcript_version -i /workspace/germline/WGS_Norm_HC_calls_recalibrated.PASS.vcf -o /workspace/germline/WGS_Norm_HC_calls_recalibrated.PASS.vep.vcf --force_overwrite
#Output tabular VEP
vep --cache --dir_cache /opt/vep_cache --dir_plugins /opt/vep_cache/Plugins --fasta /opt/vep_cache/homo_sapiens/93_GRCh38/Homo_sapiens.GRCh38.dna.toplevel.fa.gz --fork 8 --assembly=GRCh38 --offline --tab --plugin Downstream --everything --terms SO --pick --coding_only --transcript_version -i /workspace/germline/WGS_Norm_HC_calls_recalibrated.PASS.vcf -o /workspace/germline/WGS_Norm_HC_calls_recalibrated.PASS.vep.tsv --force_overwrite
Explore the VEP summaries
To load the VEP summaries, in your browser, navigate to a URL like this (don’t forget to substitute your number for #):
- http://s#.pmbio.org/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vep.vcf_summary.html
- http://s#.pmbio.org/germline/Exome_Norm_HC_calls.filtered.PASS.vep.vcf_summary.html
You should see various summary tables and graphics such as the following breakdown of variant consequences.
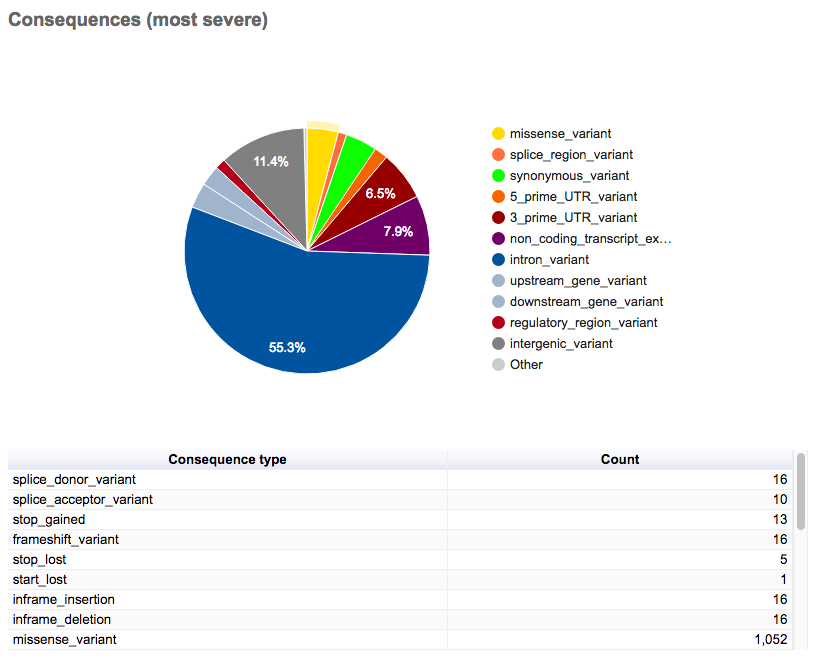
- How many variants were processed by VEP?
- What is the most common kind of variant and variant consequence?
Next steps
Note: We will continue exploring the annotated germline variants created above in a later section on germline variant interpretation, specifically in a clinical context. But, for now let’s proceed to somatic variant calling.
Acknowledgements and citations
This analysis demonstrated in this tutorial would not be possible without the efforts of developers who wrote and maintain the GATK and VEP tools. We have also acknowledged their excellent tutorials, documentation, forums, and workshop materials wherever possible. The GATK and VEP papers are also cited below. Please remember to cite these tools in your publications when using them.
- The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. Genome Res. 2010 Sep;20(9):1297-303. doi: 10.1101/gr.107524.110. Epub 2010 Jul 19.
- A framework for variation discovery and genotyping using next-generation DNA sequencing data. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. Nat Genet. 2011 May;43(5):491-8. doi: 10.1038/ng.806. Epub 2011 Apr 10.
- From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. Curr Protoc Bioinformatics. 2013;43:11.10.1-33. doi: 10.1002/0471250953.bi1110s43.
- The Ensembl Variant Effect Predictor. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. Genome Biol. 2016 Jun 6;17(1):122. doi: 10.1186/s13059-016-0974-4.