Intro to Immunogenomic Analysis
Key concepts
- Immunogenomics, adaptive immunity, immunotherapies, personalized cancer vaccines, MHC binding prediction, neoantigen identification and prioritization
Learning objectives
- Use results from the previous sections (germline variants, somatic variants, gene/transcript expression estimates, variant allele fractions, clonality estimates, etc.) to design a personalized cancer vaccine for our hypothetical patient
- Become familiar with the pvactools software for neoantigen identification, prioritization, selection, and DNA vector design
Lecture
Introduction to Immunogenomics
Nonsynonymous coding mutations alter the amino acid sequences of endogenous proteins. Proteins are naturally processed by the immunoproteasome, and the lysed peptides are loaded into the antigen presentation complexes on the surface of the cell. Sometimes, mutant peptides due to nonsynonmyous mutations have increased affinity to the antigen presentation complex, effectively eliciting a tumor-specific adaptive immune and T cell response. These peptides are called neoantigens and can be inferred for a patient’s tumor by their genomic and somatic profiles. The interaction between neoantigens and tumor-specific T cells has led to the discovery of personalized therapeutics (e.g. personalized cancer vaccines) and mechanisms of immunotherapeutic response.
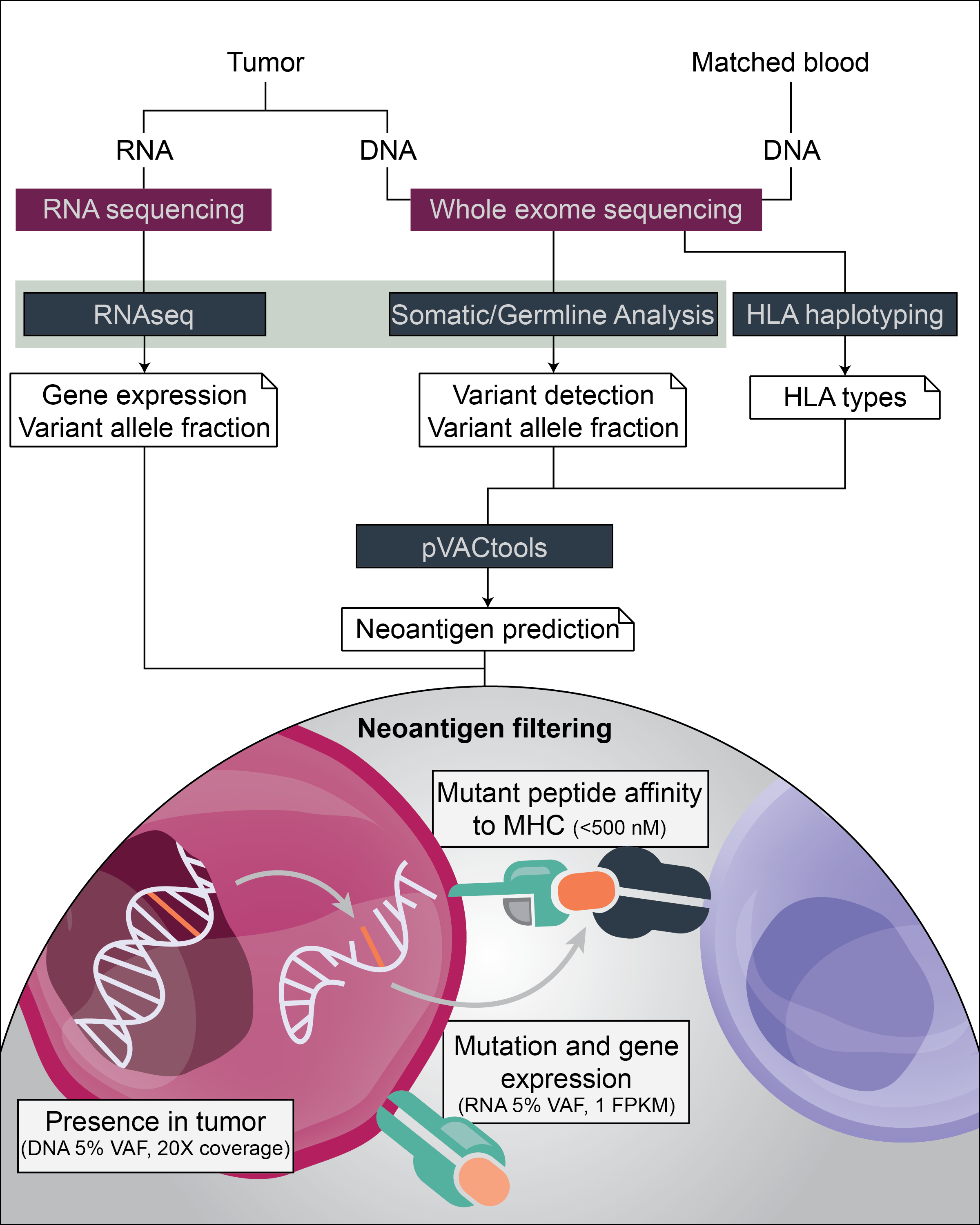
Following the identification of somatic mutations, the steps associated with identifying neoantigens include:
- Determine the normal HLA haplotypes
- Perform peptide affinity predictions
- Filter high-binding neoantigens based upon their tumor-specific expression
HLA haplotyping
The human leukocyte antigen (HLA) genes encode the major histocompatibility complex (MHC) molecules in humans. The MHC gene complex in humans is contained in a 3Mbp region on chromosome 6p21. While there are over 40 genes within this gene complex, there are nine HLA genes that are the most studied and well characterized. These genes can be very diverse across individuals, and there have been hundreds to thousands of haplotypes identified for each allele, encoding thousands of different HLA proteins. From an immunogenomics perspective, this is important because each person will present different peptides to the immune system, since different HLA proteins bind peptides and neoantigens differently.
Antigen presentation complexes are distinguished as either Class I or II. Class I complexes comprise a Class I-specific MHC molecule (encoded by HLA-A, -B, or -C) and beta-2-microglobulin (B2M), and are recognized by CD4+ T cells. Class II complexes include two MHC molecules (encoded by HLA-DPA1, DPB1, DQA1, DQB1, DRA, and DRB1), and are recognized by CD8+ T cells. Since there are nine canonical HLA genes, and each individual can have two different alleles for each gene (one each from paternal and maternal chromosomes), a person can have up two six different types of MHC-1 and 6-8 functioning MHC-II alleles.
HLA haplotyping is performed algorithmically by aligning and assembling normal DNA sequencing data to chromosome 6. There are many HLA typing tools available; however, we will be using xHLA in this workshop. One limitation of many HLA typing tools is the ability to predict both Class I and II haplotypes, since Class II HLA typing is not as well established. xHLA performs both Class I and II HLA haplotyping, allowing further characterization of both the Class I and II neoantigen landscape.
Predicting peptide affinity
The variability in the HLA gene loci results in hundreds or thousands of HLA proteins per gene, which increases the numbers of antigens that can potentially be presented by cells to the immune cells. The [Immune Epitope Database] (https://www.iedb.org/) (IEDB) has been established, summarizing experimentally determined affinity values for peptide-MHC complexes. Machine learning algorithms have been developed, trained on this rich dataset, to predict these values. In the context of cancer neoantigens, the mutant peptide affinity is used to predict how likely a peptide is to be presented to the immune cell by either the tumor or antigen presenting cells.
The inputs for these prediction algorithms include the somatic mutations of a patient’s tumor (defining the mutant peptides present in the tumor) and the matched normal HLA types, which predicts which peptide-MHC complexes may successfully be formed on the surface of cancer cells. In this workshop, we will use [pVACtools] (https://pvactools.readthedocs.io/en/latest/) to perform these predictions. The output includes the predicted affinity (in nM) between peptides and patient-specific HLA proteins. Conventionally, a binding of 500 nM or less is considered a ‘high binding’ peptide. Note that lower affinity values indicate stronger binding interactions.
Filtering neoantigens
In the next section, we will implement pVACtools to simulate the design of a personalized cancer vaccine, a therapeutic approach specifically designed for a patient based upon their tumor mutational profile. To narrow down the list of predicted, high-binding neoantigens, there are several questions to take into consideration:
-
How specifically can a tumor be seen by the immune system? pVACtools outputs the predicted binding affinity of both the mutant peptide, and the corresponding wildtype peptide. One filter includes removing neoantigens with similar ratios between mutant and wildtype peptides. Immunologically, this indicates that the immune system may have already seen the wildtype protein during development, and a T cell response to this peptide has been negatively selected from the patient’s immune system through central tolerance. Clinically, this may indicate that the wildtype peptide sequence is equally or more likely to be seen by the immune system, which could result in eliciting immune responses to normal cells (i.e. autoimmunity).
-
How likely is the mutant peptide to be expressed by the tumor? pVACtools integrates both variant coverage and expression information to further distinguish which neoantigens are most likely to be expressed by the tumor. Setting minimum coverage and VAF values in the tumor DNA positively selects for mutations present in a higher fraction of tumor cells. By targeting neoantigens in the founding clone of the tumor, it is more likely to elicit an immune response to a higher fraction of the tumor. Choosing minimum expression values in the RNA (i.e. VAF, FPKM), we are selecting mutations that are more likely to be expressed at the RNA (and potentially protein) level.