Somatic CNV Filtering/Annotation/Review
Now that we have our copy number results lets work on annotating and interpreting the calls. In this section we’ll focus on the cnvkit wgs results however the general methodology described here will be applicable to any segmented CN calls. We’ve already downloaded a gtf file ref_transcriptome.gtf containing gene annotations and their respective coordinates let’s go ahead and take advantage of this file and intersect our gene annotations with the copy number segments.
To start lets go ahead and convert our .gtf file to a .bed file with gene annotations tacked on to the end. Remeber that a .gtf is 1-based so to convert to 0-base coordinates we will have to subtract 1 from the start.
# convert the .gtf file to a .bed with gene annotations
cat /workspace/inputs/references/transcriptome/ref_transcriptome.gtf | grep -w gene | tr ";" "\t" | cut -f 1,4,5,11 | tr -d "\"" | tr -d "gene_name " | awk '{print $1"\t"$2-1"\t"$3"\t"$4}' > /workspace/inputs/references/transcriptome/gene_annotation.bed
We also will need to convert our segmented CN calls to a 0-based format as the calls are currently 1-based. We can use awk to achieve this goal and we remove the header with tail.
# change to the cnvkit wgs directory
cd /workspace/somatic/cnvkit_wgs
# convert the segmented cn calls to 0-base coord
tail -n +2 WGS_Tumor_merged_sorted_mrkdup_bqsr.cns | awk '{print $1"\t"$2-1"\t"$3"\t"$4"\t"$5"\t"$6"\t"$7"\t"$8}' > WGS_Tumor_merged_sorted_mrkdup_bqsr.2.cns
Now that we have our two pseudo bed files we can use bedtools intersect to intersect our genes with the CN output. We use the -wa and -wb options to tell bedtools we want it to print the entirety of both files we are intersecting.
bedtools intersect -wa -wb -b /workspace/inputs/references/transcriptome/gene_annotation.bed -a WGS_Tumor_merged_sorted_mrkdup_bqsr.2.cns > WGS_Tumor_merged_sorted_mrkdup_bqsr.2.annotated.cns
With our CN calls now intersected with our gene list let’s go ahead and find all CN deletions with a log2 ratio below 1 on chromosome 6 and figure out how many genes are involved in these deletions.
cat WGS_Tumor_merged_sorted_mrkdup_bqsr.2.annotated.cns | grep chr6 | awk '{if($5 < -1) print $0}' | cut -f 12 | sort | uniq | wc -l
As a final exercise let’s see what one of the more drastic CN segmented regions look like in IGV.
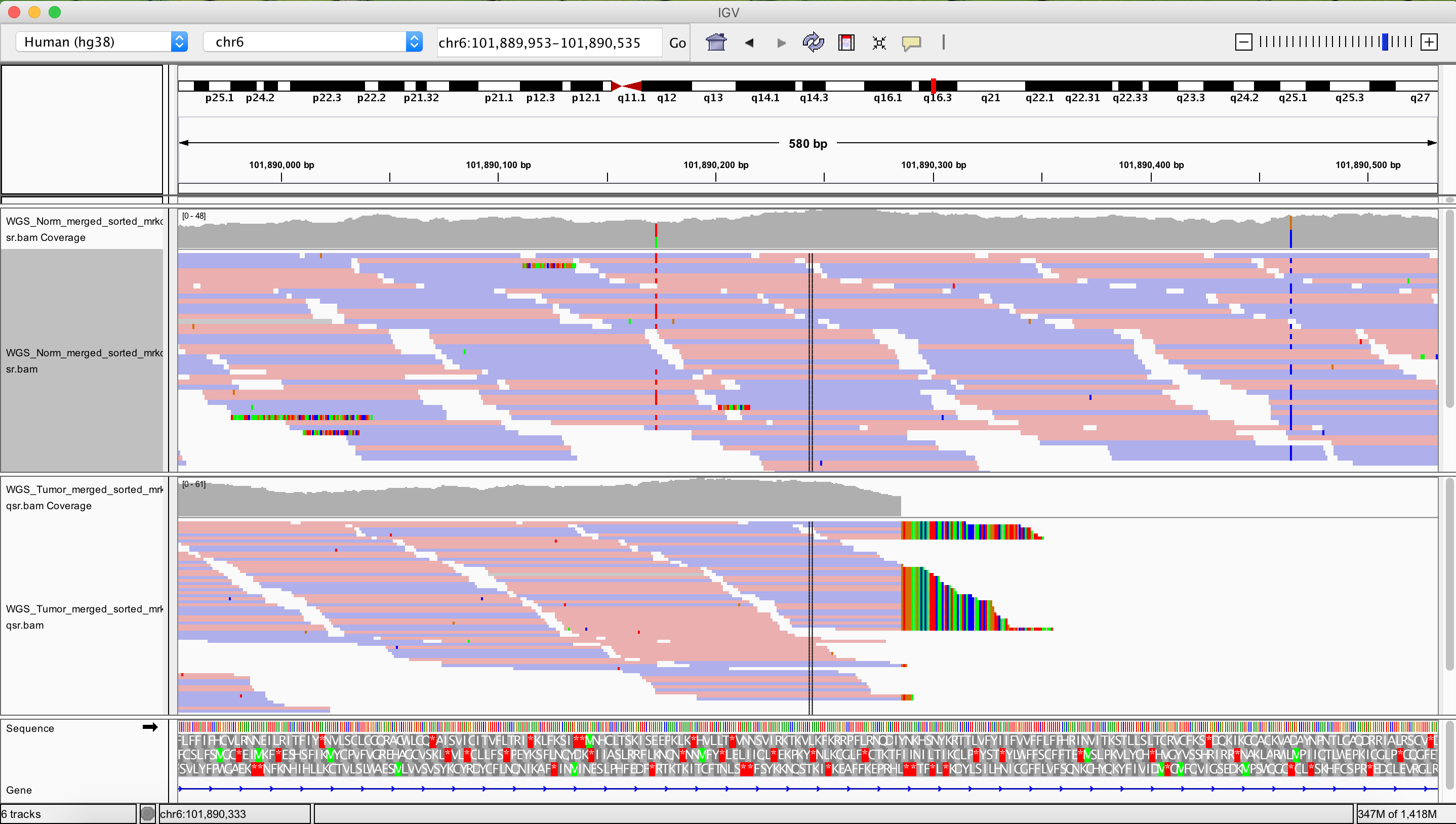
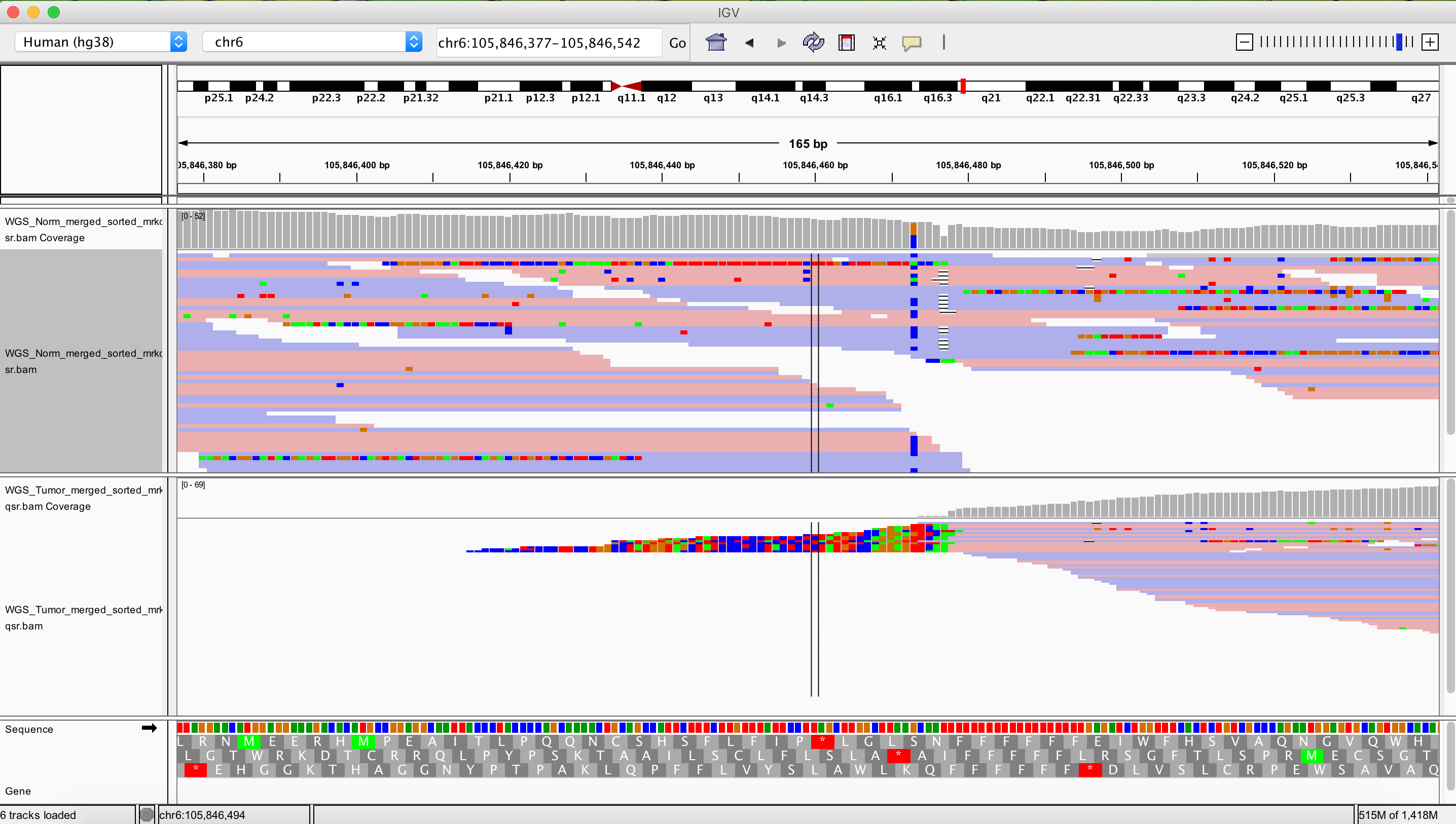
TODO: Right side breakpoint of that one is around here. chr6:105,846,162-105,847,004 Left side looks really nice as well: chr6:101,889,953-101,890,535