Germline Variant Interpretation
In this module, we will pick up where we left off at the end of Module 4. We had completed germline variant calling of exome and WGS data, performed hard- and VQSR- quality filtering, and applied VEP annotations. Now, we will attempt to filter further to prioritize variants of potential clinical significance.
Filter VEP annotated variants further for potential clinical relevance
We will use the filter_vep tool to prioritize clinically interesting variants.
The Ensembl filter_vep tool is run with the following options:
- –format vcf specifies VCF as the input file format
- -i specifies the path to input vcf file
- -o specifies the path to output vcf file
- –filter specifies the filter expression to apply (see below)
-
–force_overwrite specifies to force the overwrite of an existing file without warning
- Note - there is a difference between the information available for filtering in VEP VCF output vs VEP tabular output. Namely some VCF fields (e.g., FILTER) are not included in the tabular output. Therefore, if you wish to use filter_vep on tabular output (for ease of reading) make sure to complete any vcf-specific filtering first (e.g., using GATK SelectVariants). Alternatively, it may be possible to proceed with filtering on the VCF and then reannotate with VEP specifying tabular output to convert. Yet another option would be to use VCF annotation tools (vatools.org).
- Note - the filter_vep tool immediately and automatically limits to variants with a CSQ entry when filtering VCFs. Keep in this mind if you have annotated your VCF with the coding_only option which only adds CSQ entries for coding region alterations.
- Note - I’m obtaining different results depending on the order of filters supplied if using separate “–filter” options. This should not be the case. Combining into a single expression seems to work though.
First, let’s filter the hard-filtered exome VCF for variants of potential clinical relevance
#Filter hard-filtered exome results for clinical relevance
#Filter VEP VCF
cd /workspace/germline
filter_vep --format vcf -i /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vep.vcf -o /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vep.interesting.vcf --filter "(MAX_AF < 0.001 or not MAX_AF) and ((IMPACT is HIGH) or (IMPACT is MODERATE and (SIFT match deleterious or PolyPhen match damaging)))" --force_overwrite
The filter expression we have specified in the above example is “(MAX_AF < 0.001 or not MAX_AF) and ((IMPACT is HIGH) or (IMPACT is MODERATE and (SIFT match deleterious or PolyPhen match damaging)))”. Let’s break down this expression. There are two main types of filtering being applied with an AND operator, meaning that both must be true in order for a variant to pass filtering.
- “(MAX_AF < 0.001 or not MAX_AF)” specifies that the highest allele frequency observed in any population from 1000 genomes, ESP or gnomAD must be less than 0.001 or not observed at all. These are population allele frequencies and represent how common a variant allele is in the general/healthy population. For a variant to be considered pathogenic it should generally not be present commonly in the population, as damaging variants are expected to have been negatively selected over evolutionary time. Pathogenic cancer variants will generally be private to an affected individual or family, or at least appear only very rarely in the general population.
- ”((IMPACT is HIGH) or (IMPACT is MODERATE and (SIFT match deleterious or PolyPhen match damaging)))” specifies that a variant should have high impact on gene function (generally truncating mutations) or if only moderate function (e.g., missense mutations) then should also be considered deleterious by SIFT or damaging by PolyPhen. Note, that by using the
matchoperator we will include variants that are either deleterious or likely deleterious for SIFT and similar partial matches for PolyPhen.
To list available fields in VCF/TSV available for filtering, try the following. The meaning of most fields can be determined by reading the VEP options or filter_vep documentation.
filter_vep --list --format vcf -i /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vep.vcf
The above filtering strategy can be applied to all of the VEP annotated TSV and VCFs files that we produced for exome/WGS with either hard or VSQR quality filtering.
#Filter tabular VEP
cd /workspace/germline
filter_vep --format tab -i /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vep.tsv -o /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vep.interesting.tsv --filter "(MAX_AF < 0.001 or not MAX_AF) and ((IMPACT is HIGH) or (IMPACT is MODERATE and (SIFT match deleterious or PolyPhen match damaging)))" --force_overwrite
#Filter hard-filtered WGS results for clinical relevance
#Filter VEP VCF
filter_vep --format vcf -i /workspace/germline/WGS_Norm_HC_calls.filtered.PASS.vep.vcf -o /workspace/germline/WGS_Norm_HC_calls.filtered.PASS.vep.interesting.vcf --filter "(MAX_AF < 0.001 or not MAX_AF) and ((IMPACT is HIGH) or (IMPACT is MODERATE and (SIFT match deleterious or PolyPhen match damaging)))" --force_overwrite
#Filter tabular VEP
filter_vep --format tab -i /workspace/germline/WGS_Norm_HC_calls.filtered.PASS.vep.tsv -o /workspace/germline/WGS_Norm_HC_calls.filtered.PASS.vep.interesting.tsv --filter "(MAX_AF < 0.001 or not MAX_AF) and ((IMPACT is HIGH) or (IMPACT is MODERATE and (SIFT match deleterious or PolyPhen match damaging)))" --force_overwrite
#Filter VQSR-filtered exome results for clinical relevance
#Filter VEP VCF
filter_vep --format vcf -i /workspace/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vep.vcf -o /workspace/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vep.interesting.vcf --filter "(MAX_AF < 0.001 or not MAX_AF) and ((IMPACT is HIGH) or (IMPACT is MODERATE and (SIFT match deleterious or PolyPhen match damaging)))" --force_overwrite
#Filter tabular VEP
filter_vep --format tab -i /workspace/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vep.tsv -o /workspace/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vep.interesting.tsv --filter "(MAX_AF < 0.001 or not MAX_AF) and ((IMPACT is HIGH) or (IMPACT is MODERATE and (SIFT match deleterious or PolyPhen match damaging)))" --force_overwrite
#Filter VQSR-filtered WGS results for clinical relevance
#Filter VEP VCF
filter_vep --format vcf -i /workspace/germline/WGS_Norm_HC_calls_recalibrated.PASS.vep.vcf -o /workspace/germline/WGS_Norm_HC_calls_recalibrated.PASS.vep.interesting.vcf --filter "(MAX_AF < 0.001 or not MAX_AF) and ((IMPACT is HIGH) or (IMPACT is MODERATE and (SIFT match deleterious or PolyPhen match damaging)))" --force_overwrite
#Filter tabular VEP
filter_vep --format tab -i /workspace/germline/WGS_Norm_HC_calls_recalibrated.PASS.vep.tsv -o /workspace/germline/WGS_Norm_HC_calls_recalibrated.PASS.vep.interesting.tsv --filter "(MAX_AF < 0.001 or not MAX_AF) and ((IMPACT is HIGH) or (IMPACT is MODERATE and (SIFT match deleterious or PolyPhen match damaging)))" --force_overwrite
Explore the filtered results
As an example, let’s explore the exome joint genotype calls after VQSR quality filtering, VEP annotation, and VEP filtering as above. If we use the tab-delimited files we could open directly in Excel for example. In your browser, download the appropriate file using a URL like this (don’t forget to substitute your number for #):
- http://s#.pmbio.org/germline/Exome_Norm_GGVCFs_jointcalls_recalibrated.PASS.vep.interesting.tsv
The above filtering has limited the results to less than 10 germline variants of potential interest. Has the VEP filtering worked as expected? I.e., Do the variants have only low or null MAX_AF values? Do they have only MODERATE or HIGH impact? If MODERATE, do they also have deleterious or damaging SIFT/PolyPhen scores?
Exercise
Are there any variants of obvious clinical relevance to breast cancer?
Answer
Look at the CLIN_SIG column. At least one variant includes a pathogenic assessment. This variant is a nonsense (stop_gained) variant in BRCA1, a well known breast cancer predisposition gene responsible for hereditary breast cancer
Try using the variant’s Existing_variation IDs (e.g., dbSNP) to search the ClinGen Allele Registry, identify the correct allele, and then follow the links (if any) to ClinVar or other resources of interest.
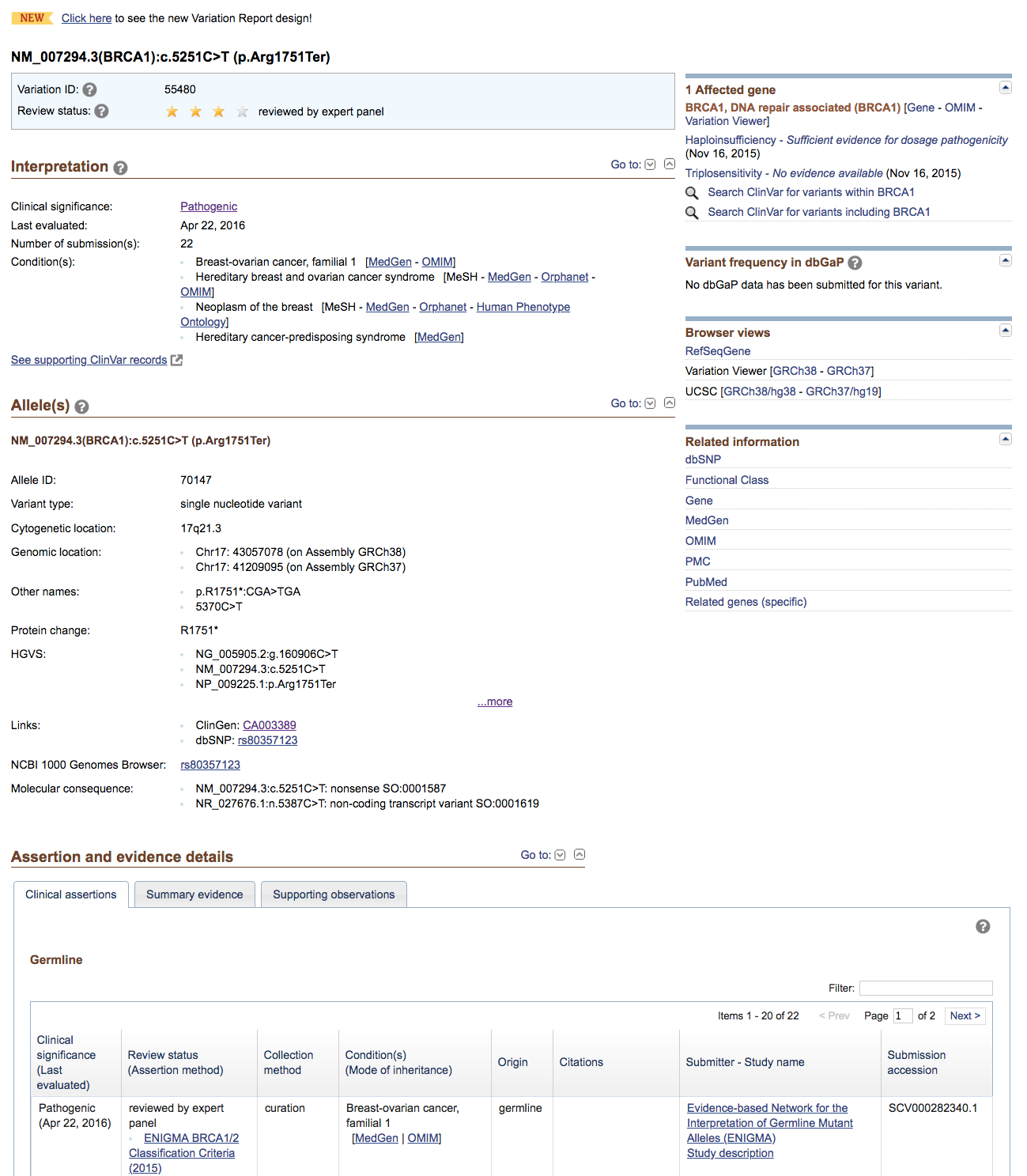
Reviewing the ClinVar record, we can see that in fact this variant has been reviewed by an expert panel (ENIGMA) who have assessed this variant as pathogenic for breast cancer. Based on this, it is quite likely that the patient had a predisposition to develop breast cancer. In fact, recall that there was actually a family history of breast cancer.
More Exercises
- Are there any variants in known cancer genes? Try intersecting the remaining genes with the Cancer Gene Census genes?
- Manually review any variants of interest in IGV
- Determine the overlap of clinically relevant variants between hard- and VQSR quality filtered inputs, between WGS and Exome. What could explain these discrepancies?
- Experiment with your own filtering strategies to increase or decrease the stringency for prioritizing “clinically relevant” variants.